

Phase I trials
Drug development trials are traditionally divided into 3 phases serving different purposes (Table 1). Phase I assessments, which represent the first-in-human evaluation of any new compound, focus on determination of the optimal dosing for further testing. Primary endpoints therefore typically include dose-limiting toxicities (DLT), the maximum tolerated dose (MTD), and the recommended phase II dose. Secondary endpoints comprise pharmacokinetics (PK), pharmacodynamics (PD), and preliminary anti-tumor activity. Patients, rather than healthy volunteers, with refractory cancer of any type are eligible.

Phase I trials consist of the dose escalation portion and the cohort expansion portion. The dose escalation part is dedicated to assessment of PK, safety and MTD. This segment usually encompasses only very few patients for each of several dose levels. In the expansion phase, a greater number of patients is recruited into one or two selected dose levels with the purpose of decreasing confidence intervals. Adverse events (AEs) of cytotoxic chemotherapy usually become evident within the very first treatment cycles.
Changes brought about by the introduction of targeted treatments
The principles of designing oncology phase I trials have changed in the context of targeted therapies, which nowadays constitute the bulk of investigational drugs. Targeted agents need to be administered for longer periods, often without the frame of defined cycles. Also, toxicity follows a different pattern. According to a review of 36 phase I trials conducted with targeted therapies, the majority of severe AEs only becomes apparent after cycle 1, and 50 % of patients experience their worst-grade toxicity after completion of the customary DLT assessment period [1]. Moreover, targeted therapies usually show no dose-response-relationship in the phase I setting, which renders determination of the ideal dose difficult.
All things considered, MTD and DLT might be concepts of the past that no longer apply to modern drug development, and their definitions need to be reconsidered. Another aspect is the choice of PD markers in the targeted era, as the optimum target inhibition, and thus the optimum sample size, is usually unknown. Mostly, tumor tissue is used to determine PD markers.
A major difference between phase I studies for cytotoxic drugs and those for targeted compounds relates to the amount of early efficacy data. With chemotherapy, the focus used to be on safety rather than on anti-tumor activity, and as several types of tumors were treated, conclusions about certain types of tumor that responded particularly well were difficult. Many targeted agents that have been tested over the last 10 to 15 years, however, showed response rates of more than 50 % in the phase I setting. This development has become possible due to the improved understanding of tumor biology. At present, many oncology phase I studies are no longer involving patients with any cancer type, but instead those who appear most suitable for the new drug.
Advantages of multiple expansion cohorts
All of these changes have led to considerable increases in the size of cohorts involved in phase I trials, the number of expansion cohorts, and the objectives of the studies (Table 2). A typical example is immunotherapy trials that tend to comprise 6–8 or even more expansion cohorts, which enables the testing of different doses and schedules in different tumor types. According to an analysis by Mullard et al., recent industry-sponsored phase I trials on immunotherapies included as many as 1,000 to 2,000 patients, thus drawing level with the traditional size of cardiovascular studies [2].

The inclusion of multiple expansion cohorts in phase I trials allows for simultaneous assessment of drug activity and evaluation of predictive biomarkers across various tumor types as well as further cohort expansion when promising efficacy signals occur. Also, multiple expansion cohorts lead to rapid enlargement of safety databases and speed up the drug development process for clearly efficacious drugs, which thus can benefit patients early on.
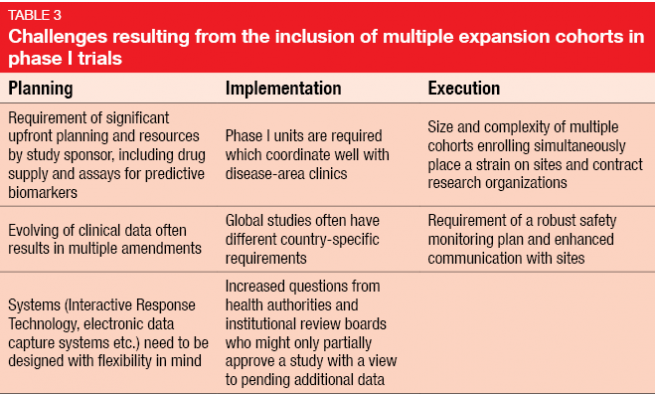
Challenges due to multiple expansion cohorts
On the other hand, there is a number of unique complexities and challenges with respect to planning, implementation and execution (Table 3). Protocols must be amended due to emerging data, sometimes repeatedly. An example of this is the KEYNOTE-001 trial [3] evaluating pembrolizumab in melanoma that underwent eight amendments, in the course of which the total sample size rose from 32 to 1.067 patients. Amendments often need to be implemented at different sites across the world, as these huge phase I trials are frequently conducted globally. In contrast, classical phase I studies used to be restricted to one site.
Concerns have been uttered that the changes in design and objectives will hamper the quality of safety data obtained in phase I. In their article on seamless oncology-drug development published in the New England Journal of Medicine in 2016, Powell et al. posed a range of questions regarding the design of large first-in-human cancer trials (Table 4) [4].


REFERENCES
- Postel-Vinay S et al., Phase I Trials of Molecularly Targeted Agents: Should We Pay More Attention to Late Toxicities? J Clin Oncol 2011; 29(13): 1728-1735
- Mullard A, 2016 FDA drug approvals. Nat Rev Drug Discov 2017; 16(2): 73-76
- Robert C et al., Three-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol 2016; 34 (suppl; abstr 9503)
- Prowell TM et al., Seamless oncology-drug development. N Engl J Med 2016; 374: 2001-2003
Author: Chia-Chi (Josh) Lin
© 2017 Springer-Verlag GmbH, Impressum