

Management of patients with relapsed/refractory CLL: what is new?
VISION: stop-start approach with ibrutinib/venetoclax
The optimal novel-agent approach for patients with chronic lymphocytic leukemia (CLL) is subject to research. Targeted therapies have become the undisputed standard of care in both relapsing/refractory and treatment-naïve settings. The choice of regimen remains, however, disputable. Continuous BTK inhibition confers the risk of cumulative toxicity and acquired resistance, while time-limited combination therapies may result in relatively high adverse event (AE) rates and lead to overtreatment of patients with favorable risk. Discontinuation of treatment based on the depth of response constitutes a possible strategy. Furthermore, most of the randomized comparisons are conducted with the first-generation BTK inhibitor ibrutinib that is potentially cardiotoxic. Therefore, this is not an optimal drug for the elderly, comorbid population.
In the setting of relapsed/refractory CLL, the phase II VISION HO141 trial assessed the feasibility of observation alone in patients who had achieved undetectable minimal residual disease (uMRD) after 15 cycles of ibrutinib plus venetoclax [1]. This strategy was compared to ibrutinib maintenance following the combination period. Once patients in the observation arm showed CLL progression, the combined treatment was reinitiated. Progression was determined according to the iwCLL criteria or MRD > 10-3 plus MRD > 10-2 at least 1 month later. Venetoclax treatment after progression was limited to 12 months, while ibrutinib was administered until progression. The primary endpoint was the progression-free survival (PFS) rate 12 months after treatment discontinuation in the observation arm.
Freedom from progression in 98 %
Among 197 patients who received the initial combination regimen and underwent MRD assessment by flowcytometry, 72 developed uMRD, while MRD remained detectable in 125 individuals. The latter were treated with ibrutinib maintenance in a non-randomized manner. In the uMRD group, 24 and 48 patients were randomized to ibrutinib maintenance and observation, respectively.
The primary endpoint of the trial was met. One year after treatment discontinuation, 98 % of patients in the observation arm were progression-free. At that time, 71 % had uMRD (< 10-4) in the peripheral blood (Figure 1). In patients without uMRD who had been assigned to non-randomized ibrutinib maintenance, MRD levels have remained stable for one year. Overall survival (OS) rates at 27 months were 100 %, 98 % and 92 % for patients on ibrutinib maintenance, observation, and non-randomized ibrutinib maintenance.
Seven patients in the observation arm reinitiated combined treatment with ibrutinib and venetoclax upon MRD positivity; six of these achieved de-novo complete remission within 3 cycles, and the 7th patient was awaiting evaluation at the time of the analysis. AE rates naturally decreased in the observation arm compared to the other patient groups after cycle 15. The authors concluded that an MRD-guided stop-start strategy of ibrutinib/venetoclax is feasible in patients with relapsed/refractory CLL and can be recommended.
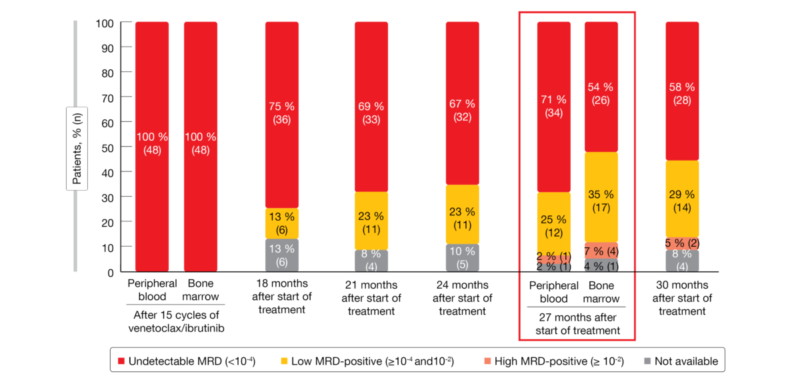
Figure 1: Minimal residual disease development after the end of fixed-duration treatment with 15 cycles of ibrutinib/venetoclax
MRD-driven triple therapy
Roeker et al. conducted a phase II study with the aim of identifying a subset of patients responding to ibrutinib monotherapy who have persistent MRD and might benefit from a combined strategy [2]. After ≥ 6 months of ibrutinib treatment in any line, the PI3Kδ inhibitor umbralisib and the anti-CD20 antibody ublituximab (U2) were added to ibrutinib in patients with detectable MRD. Treatment continued until they achieved uMRD in the peripheral blood on two sequential occasions. Overall, the triplet therapy was administered for a maximum of 24 cycles until the initiation of treatment-free observation regardless of MRD status. Durability of remission following treatment discontinuation was monitored using sequential MRD assessments. The study design permitted retreatment with ibrutinib and U2 upon clinical progression after ≥ 6 months of treatment-free observation. Twenty-eight and 27 patients were evaluable for safety and efficacy, respectively. Two thirds and one third had received ibrutinib in the first line and for relapsed/refractory disease, respectively. The uMRD rate was defined as the primary endpoint.
This was the first non–venetoclax-containing MRD-driven, time-limited approach utilizing the combination of BTK and PI3Kδ inhibitors with an anti-CD20 antibody. It gave rise to deep remissions, with a uMRD rate of 77 % and median time to first uMRD of 7.4 months. Only 4 % of patients completed 24 cycles of ibrutinib plus U2 without achieving MRD negativity, and 19 % remained on therapy with detectable MRD at the time of the analysis. PFS from study entry and from entering treatment-free observation was excellent, with only one progression event.
The AE profile of ibrutinib remained unchanged after the addition of U2. Most AEs observed in the study were rated as low-grade. Two patients discontinued all therapy due to AEs including rash and arthralgia; both had uMRD at that time and were able to enter treatment-free observation. Overall, this add-on approach for patients on continuous ibrutinib allowed for tailored, time-limited therapy and sustained treatment-free observation. The study continues to enroll, with other cohorts exploring the addition of U2 to acalabrutinib or venetoclax.
Three-year follow-up of ASCEND
Compared to ibrutinib, the next-generation BTK inhibitor acalabrutinib is more selective and has decreased alternative-target activity in vitro [3, 4]. According to the primary analysis of the phase III ASCEND trial after a median follow-up of 16.1 months, acalabrutinib monotherapy exhibited superior PFS and a favorable safety profile compared with physicians’ choice, i.e., idelalisib/rituximab (IdR) or bendamustine/rituximab (BR) in patients with relapsed/refractory CLL after ≥ 1 systemic therapy [5]. Both the acalabrutinib arm and the IdR/BR arm contained 155 patients. As most of the doctors chose IdR (118 of 153 patients in the comparator arm), the study was in fact the first randomized comparison of BTK and PI3K inhibitors in relapsed/refractory CLL.
The updated results at 3 years of follow-up presented at ASH 2021 by Jurczak et al. showed maintained efficacy and safety of acalabrutinib that was favorable compared to the standard-of-care regimens [6]. Median PFS for the experimental arm had not been reached yet, while this was 16.8 months in the control arm (HR, 0.29; p < 0.0001). At 36 months, the PFS rates amounted to 63 % vs. 21 %. Within the control population, patients treated with IdR fared better than those receiving BR; the 36-month PFS rates were 25 % vs. 9 % for IdR and BR, respectively. Acalabrutinib reduced the risk of progression or death by 69 % and 75 %, respectively (p < 0.0001 each). The advantage conferred by the novel BTK inhibitor held true irrespective of the presence of high-risk genetic features. In patients without and with del(17p), median PFS had not been reached on acalabrutinib treatment, while this was 20.3 and 13.8 months, respectively, in the control arm (Figure 2). A similar picture emerged with respect to IGHV mutation status. The PFS results favored acalabrutinib in all subgroups (e.g., number of prior therapies, presence of bulky disease, Rai stage at screening). Median OS had not been reached in either arm. At 36 months, 80 % vs. 73 % of patients were alive despite the crossover offered to 76 patients progressing in the control arm. Also, the overall response rates did not differ between acalabrutinib and IdR/BR (83 % vs. 85 %; p = 0.62).
The longer-term follow-up did not reveal any new safety findings in the experimental arm. Acalabrutinib maintained an acceptable tolerability profile, and fewer patients discontinued treatment due to AEs despite longer exposure. As the authors noted, these data support the use of acalabrutinib in patients with relapsed/refractory CLL including those with high-risk features.
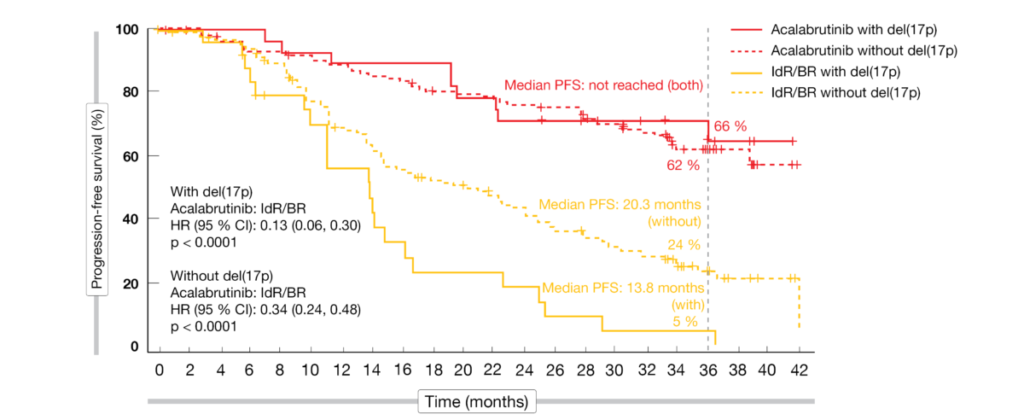
Figure 2: ASCEND: acalabrutinib vs. idelalisib/rituximab and bendamustine/rituximab according to del(17p) status
Acalabrutinib:AE and Q-TWiST analyses
The randomized, phase III ELEVATE-RR study compared acalabrutinib with ibrutinib, demonstrating non-inferior PFS and improved tolerability in pretreated patients with del(17p) and/or del(11q) [7]. Seymour et al. conducted a post-hoc analysis of the study to further characterize the BTK-inhibitor–associated AEs and the safety profile of acalabrutinib [8]. Event-based analyses demonstrated a higher BTK-inhibitor–related toxicity burden with ibrutinib. Acalabrutinib showed comparatively lower incidence, exposure-adjusted incidence, and exposure-adjusted time spent with cardiovascular-related toxicities including atrial fibrillation/flutter, hypertension, and bleeding. Moreover, cumulative incidences of hypertension and atrial fibrillation/flutter were lower with acalabrutinib in patients without a history of these events.
Based on the ASCEND and ELEVATE-RR trials, a quality-adjusted time without symptoms or toxicity (Q-TWiST) analysis assessed the quality-adjusted survival benefits of acalabrutinib versus the comparator agents [9]. The results reported at ASH 2021 indicated significant quality-adjusted survival benefit of acalabrutinib compared with idelalisib or bendamustine plus rituximab. Compared with ibrutinib, survival gains varied by toxicity definition, but were numerically higher in all analyses.
Pirtobrutinib after BTK pretreatment
The benefits achieved with covalent BTK inhibitors are often diminished due to resistance [10]. Available subsequent treatment options include BCL2 inhibitors, combination regimens and, last but not least, third-generation BTK inhibitors. The potent non-covalent BTK inhibitor pirtobrutinib shows > 300-fold selectivity for BTK vs. 370 other kinases and favorable pharmacologic properties [11].
Mato et al. reported the findings achieved with pirtobrutinib in BTK-pretreated CLL/SLL patients enrolled in the phase I/II BRUIN study [12]. In 252 efficacy-evaluable individuals, the overall response rate amounted to 68 % and remained stable over time. Pirtobrutinib was effective irrespective of the presence of high-risk genetic aberrations, the type of pretreatment, and the reason for prior BTK inhibitor discontinuation (Figure 3). Median PFS in patients who were at least BTK-inhibitor–pretreated (median number of prior lines, 3) had not been reached yet; in those who had at least received BTK and BCL2 inhibitors (median number of prior lines, 5), median PFS was 18 months. Moreover, the treatment proved efficacious in patients failing previous chemotherapy, monoclonal antibodies, covalent BTK inhibitors, BCL2 inhibitors, and PI3K inhibitors (i.e., the so-called “penta failures”).
The favorable safety and tolerability profile observed in the study was consistent with the design of pirtobrutinib as a highly selective and non-covalent reversible BTK inhibitor. No dose-limiting toxicities were reported, and the maximum tolerated dose had not been reached. AEs of special interest included bruising, rash, and arthralgia. In their conclusion, the authors summarized that pirtobrutinib demonstrates promising efficacy in CLL/SLL patients previously treated with BTK inhibitors. Randomized global, phase III trials including BRUIN CLL-321, BRUIN CLL-322 and BRUIN CLL-313 are currently evaluating this agent in the setting of CLL/SLL.
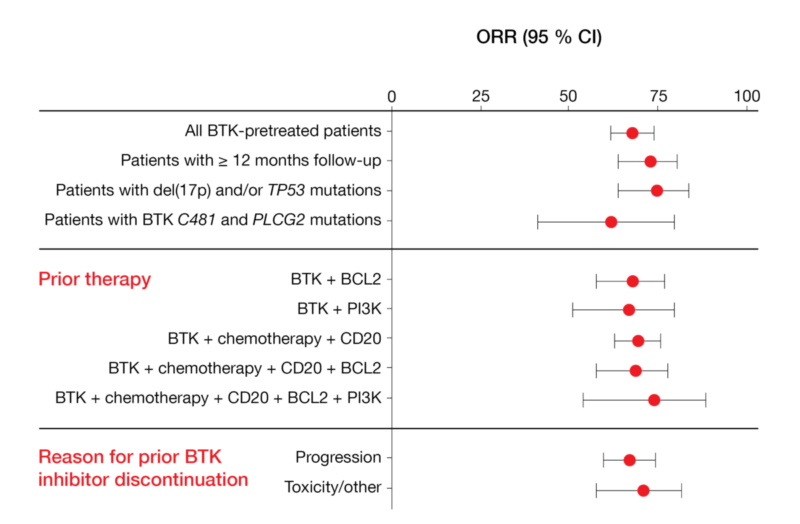
Figure 3: Responses obtained with pirtobrutinib irrespective of patient and pretreatment characteristics
MK-1026
Resistance against BTK inhibitors develops primarily through mutations at the cysteine binding site (C481) or PLCγ2 [13, 14]. MK-1026 has been developed as a non-covalent, potent inhibitor of both wild-type and C481S-mutated BTK [15]. The preliminary recommended phase II dose was determined at 65 mg/d in a phase I/II dose escalation and expansion study performed in patients with hematologic malignancies. At ASH 2021, Woyach et al. presented the efficacy and safety of MK-1026 65 mg/d in study participants with CLL/SLL treated during the dose expansion phase [16].
MK-1026 was shown to have promising anti-tumor activity, with a manageable safety profile. Among 38 efficacy-evaluable patients, 57.9 % responded to treatment. Median duration of response had not been reached yet at the time of the analysis. Almost 94 % achieved decreases in tumor volume, and decreases ≥ 50 % were seen in 69.7 %. Responses occurred in heavily pretreated patients and in those progressing on prior covalent BTK inhibition.
The most common AEs comprised fatigue (33.1 %), constipation (31.4 %), and dysgeusia (28.0 %). Grade ≥ 3 AEs predominantly included hypertension (9.3 %) and fatigue (3.4 %). Further evaluation of MK-1026 in B-cell malignancies at doses of ≥ 65 mg is ongoing.
REFERENCES
- Niemann CU et al., Time-limited venetoclax and ibrutinib for patients with relapsed/refractory chronic lymphocytic leukemia who have undetectable minimal residual disease – primary analysis from the randomized phase 2 VISION HO141 trial. ASH 2021, abstract 69
- Roeker LE et al., A phase 2 study evaluating the addition of ublituximab and umbralisib (U2) to ibrutinib in patients with chronic lymphocytic leukemia (CLL): a minimal residual disease (MRD)-driven, time-limited approach. ASH 2021, abstract 395
- Barf T et al., Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther 2017; 363(2): 240-252
- Byrd JC et al., Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med 2016; 374(4): 323-332
- Ghia P et al., ASCEND: Phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol 2020; 38(25): 2849-2861
- Jurczak W et al., Acalabrutinib vs. rituximab plus idelalisib or bendamustine in relapsed/refractory chronic lymphocytic leukemia: a three-year follow-up of the ASCEND trial. ASH 2021, abstract 393
- Byrd JC et al., Acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia: results of the first randomized phase III trial. J Clin Oncol 2021; 39(31): 3441-3452
- Seymour JF et al., Characterization of Bruton tyrosine kinase inhibitor-related adverse events in a head-to-head trial of acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia. ASH 2021, abstract 3721
- Seymour JF et al., A quality-adjusted survival (Q-TWiST) analysis to assess benefit-risk of acalabrutinib versus idelalisib/bendamustine plus rituximab or ibrutinib among relapsed/refractory chronic lymphocytic leukemia (CLL) patients. ASH 2021, abstract 3722
- Woyach JA et al., BTKC481S-Mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol 2017; 35(13): 1437-1443
- Brandhuber BJ et al., LOXO-305, a next generation reversible BTK inhibitor, for overcoming acquired resistance to irreversible BTK inhibitors. Clin Lymphoma Myeloma Leuk 2018; 18: S216
- Mato AR et al., Pirtobrutinib, a highly selective, non-covalent (reversible) BTK inhibitor in previously treated CLL/SLL: updated results from the phase 1/2 BRUIN study. ASH 2021, abstract 391
- Woyach JA et al., Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med 2014; 370(24): 2286-2294
- Jones D et al., PLCG2 C2 domain mutations co-occur with BTK and PLCG2 resistance mutations in chronic lymphocytic leukemia undergoing ibrutinib treatment. Leukemia 2017; 31(7): 1645-1647
- Woyach JA et al., Final results of phase 1, dose escalation study evaluating ARQ 531 in patients with relapsed or refractory B-cell lymphoid malignancies. Blood 2019; 134(S1): 4298
- Woyach JA et al., Preliminary efficacy and safety of MK-1026, a non-covalent inhibitor of wild-type and C481S mutated Bruton tyrosine kinase, in B-cell malignancies: a phase 2 dose expansion study. ASH 2021, abstract 392
© 2022 Springer-Verlag GmbH, Impressum