

Phase II trials
Phase II studies provide initial assessment of efficacy in a more homogeneous patient population.Screening out of ineffective drugs is an objective here, as is the identification of promising new agents for future evaluation. The activity of a compound is assessed in a given tumor type, which allows for selection of tumor types for further study. Also, safety (types and incidence of AEs) is further defined in a specific patient population/ disease setting.
Overall, sufficient evidence should be generated to support the phase III development of the compound. As part of the strategic development, the phase II trial sets the course for the conduct of the phase III trial in a certain disease or, in case of negative results, demonstrates its futility.
Types of phase II studies
Phase II studies can be investigator-initiated or based on co-operative group efforts. They are conducted in the light of a clinically driven rationale/ unmet need and usually evaluate single agents or combinations with existing therapies. The consensus recommendations on the design of phase II clinical trials testing cancer therapeutics by the Clinical Trial Design Task Force of the National Cancer Institute Investigational Drug Steering Committee details several types of phase II trials (Figure 1) [1].
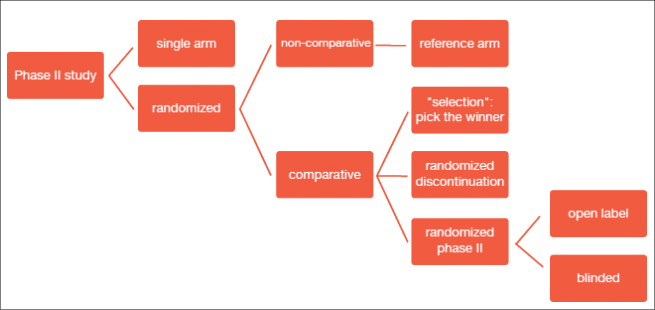
Figure 1: Types of phase II studies
In clinical practice, single-arm versus randomized studies have been used as the primary categorization. Single-arm studies include a smaller sample size and a one-stage or two-stage design. They are conducted with the expectation of certain response rates based on historical information or control databases. Randomized trials, on the other hand, are generally larger and ideal for the comparison of a primary endpoint or for “calibration” against a control arm, where expected outcomes are less certain. They can have a comparative or non-comparative design. Randomized trials are more expensive than single-arm studies, but offer the advantage of exploring multiple arms at once.
Population selection
It is important to include patients who are most likely to benefit from the intervention being tested. Those who are unlikely to benefit or who show a greater risk of harm should be excluded. Overall, a homogeneous population should be strived for. Selection can be driven by a priori information or clinical factors. A priori information relates to knowledge on the disease prevalence of a particular protein or gene abnormality that predicts for a greater benefit based on the mode of action of a specific drug (biological rationale); also, existent pre-clinical evidence for activity of a drug/ proof of concept in a distinct tumor type might be decisive. Clinical factors include responses observed in the phase I setting, or the biological rationale in a disease area of unmet need.
Population enrichment follows a two-stage design [2]. The learn phase comprises determination of the biomarker status and comparison of the study drug with the comparator using two cohorts that consist of biomarker-positive and biomarker-negative patients. In the confirm phase, the study drug is tested against the comparator in the biomarker-positive population only, if a clear trend of superior efficacy versus the biomarker-negative cohort has been demonstrated. If similar treatment effects occurred in both cohorts, the entire population can be carried forward.
Pertinent endpoints
A number of study endpoints to choose among has been defined (Table 1). For primary endpoint selection, the decision between response rates (RR) and progression-free survival (PFS) requires understanding of the expected drug effects on the disease (e. g., cytotoxic versus cytostatic activity, first line versus second line versus maintenance). The rigor of the choice between RR and PFS according to Response Evaluation Criteria In Solid Tumors (RECIST) depends on the goal of the study, e. g., activity “screening” trial (investigator review) or decision-making “go/ no-go” trial (independent review).
When RR has been selected for a monotherapy trial, a single-arm design is acceptable. Randomization is required for testing of different doses or schedules or for the comparison with other active therapies. For the assessment of combination therapy (e. g., standard therapy with or without a novel agent, or combinations of novel agents), a randomized design is usually preferred.
Adaptive trial designs
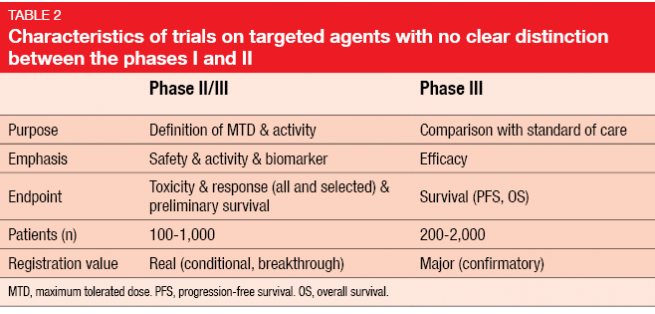
With the dawning of the era of targeted treatments, the differences between the classical phases of drug development have increasingly become blurred. In the early stage, there is often no clear distinction between phases I and II (Table 2). This amalgamation is enhanced by the possibility of accelerated approval, which is usually based on combined data from these two phases. After conditional approval has been granted by the authorities, a phase III study needs to be conducted as a confirmatory trial for full approval.
Adaptive designs also include intermixture of phases II and III. The design presented in Figure 2 shows a trial evaluating two experimental drugs alone and in combination [3]. After the single agent (drug B) has been selected in phase II, it continues into phase III. The number of patients and the randomization in phase II are chosen adaptively, and phase II results determine the sample size in phase III. Interim analyses might be used to halt phase III early on, either for futility or for expected success. There is also the possibility of omitting phase II: when the design of the phases I and II clearly answers the key questions on safety and efficacy, regulatory authorities now even consider a direct transition to phase III.
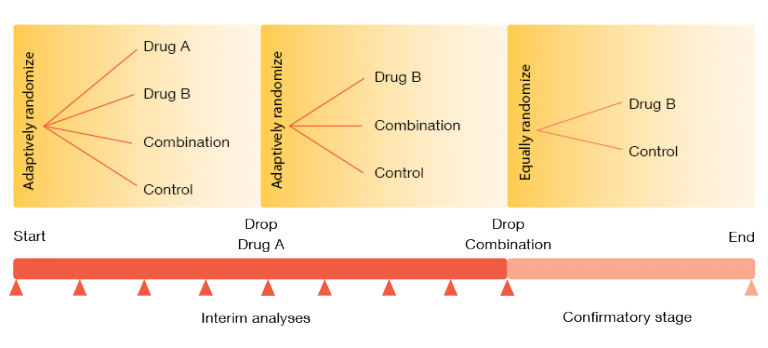
Figure 2: Example of a seamless phase II-III design

REFERENCES
- Seymour L et al., The design of phase II clinical trials testing cancer therapeutics: consensus recommendations from the clinical trial design task force of the national cancer institute investigational drug steering committee. Clin Cancer Res 2010; 16(6): 1764–1769
- Ananthakrishnan R et al., Design of oncology clinical trials: a review. Crit Rev Oncol Hematol 2013; 88(1): 144-153
- Berry DA, Adaptive clinical trials in oncology. Nat Rev Clin Oncol; 2011; 9(4): 199-207
Author: Gilberto Schwartsmann
© 2017 Springer-Verlag GmbH, Impressum