

SCLC: genomic alterations pave the way to targeted approaches
Rapid growth and early development of metastatic disease are characteristic of small-cell lung cancer (SCLC), which constitutes approximately 15 % of all lung cancer cases [1]. In limited-stage disease, a cure is possible with chemoradiotherapy. However, 68 % of patients present with extensive-stage SCLC (ES-SCLC). Although high initial responses to platinum-based chemotherapy and radiotherapy are observed, recurrence of chemo-refractory disease takes place as a rule.
At present, the lack of effective therapies for relapsed SCLC is one of the greatest unmet needs in the management of lung cancer patients. Nearly all SCLC cases are attributable to cigarette smoking, which has implications for the mutational landscape of these cases, and thus for the potential use of certain treatments.
Genomic profiling of SCLC patients
Ali et al. reviewed 883 patients with SCLC using a comprehensive genomic profiling approach [2]. Importantly, all types of genomic alterations were identified (i.e., base-pair substitutions, insertions/deletions, copy number alterations, rearrangements). This study is the largest to date that describes the genomic profiles of SCLC patients through the course of their clinical care.
The results here showed that frequent genomic alterations are present in SCLC. Genomic alterations, which included MYCL1 fusions, were consistent with those in the published literature. The most commonly altered genes were TP53 (90 %), RB1 (69 %), MLL2 (12.0 %), LRP1B (10.9 %), PTEN (8.5 %), MYCL1 (8.0 %), RICTOR (6.5 %), and MYC (6.1 %). Focal amplifications were frequent, and included RICTOR/FGF10 on chromosome 8 and MYCL1 on chromosome 1. MYCL1 amplification was found in 68 (7.8 %) of the patients. Seven patients harboured MYCL1 fusions, and five of these also had MYCL1 amplification.
These patients included the unique index case of a never-smoker whose tumour harboured JAZF1-MYCL1 without amplification of MYCL1. This patient experienced durable complete remission over 18 months when treated with the investigational aurora A kinase inhibitor alisertib (MLN8237), and durable partial response to nivolumab. The biological implication of this is that JAZF1-MYCL1 might ectopically stabilise functional MYCL1 expression, thus hyper-activating the downstream target aurora kinase, as well as hyper-inhibiting the downstream target PD-L1. MYCL1 amplifications might represent a less dramatic elevation of downstream activity, but they still confer sensitivity to aurora kinase inhibitors and PD-1 inhibitors. Therefore, some patients, and particularly those harbouring MYCL1 amplifications, might benefit from the combination of an aurora kinase inhibitor and an immunotherapeutic drug.
The tumour mutational burden (TMB) in SCLC was calculated at 9.9 mutations/ megabase. In comparison, the TMB for melanoma is 12.6 mutations/ megabase, while it is lower in other tumours. Assuming that the TMB correlates with the efficacy of PD-1/PD-L1 inhibitors, the distribution of the TMB in SCLC suggests a similar response to immunotherapy as seen in NSCLC.
Aurora A kinase inhibition plus paclitaxel
Aurora A kinase (AAK) is a key regulator of mitosis. It can be overexpressed or amplified in a range of solid tumours and haematological malignancies. Inhibition of AAK leads to disrupted mitosis and cell death, which makes AAK a potential target for anti-cancer therapies. AAK inhibitors appear to be effective in SCLC cell lines, and especially in those with amplification and/or high expression of Myc [3, 4], which is a main driving oncogene in many cancers. Myc amplification of overexpression occurs in 18 % to 31 % of SCLCs, and is more common in chemo-refractory disease [3].
The investigational, orally available, selective, small-molecule, AAK inhibitor alisertib was tested in combination with paclitaxel in a randomised phase II study [5]. Patients with SCLC participated who had previously been treated with one platinum-based chemotherapy regimen and had experienced relapses earlier than 180 days of completion. The patients in the control arm received placebo plus paclitaxel. Eighty-eight individuals were enrolled in each study arm.
Activity in c-Myc–expressing tumours
For PFS in the ITT population, which was defined as the primary endpoint, the analysis revealed a significant advantage of the alisertib combination (3.32 vs. 2.17 months; HR, 0.71; p = 0.038). Patients with resistant or refractory relapses also experienced significant PFS benefit (2.86 vs. 1.64 months; HR, 0.659; p = 0.037). For OS, DCR and ORR, the results hinted at favourable outcomes with the alisertib combination, although the differences did not reach significance.
Alisertib and paclitaxel have overlapping toxicities. Grade ≥ 3 AEs occurred more frequently with alisertib plus paclitaxel (76 % vs. 51%), as did drug-related serious AEs (32 % vs. 7 %). The most common AEs with the alisertib combination included diarrhoea, fatigue, neutropenia, anaemia and stomatitis. Neutropenia dominated among the grade ≥ 3 AEs (38 % vs. 6 %). All of the grade ≥ 3 AEs were at least two-fold more frequent in the experimental arm than in the control arm. AE-related drug discontinuation occurred more frequently in the experimental arm (15 % vs. 6%), as also seen for dose reductions due to AEs (38 % vs. 10 %).
With c-Myc protein expression believed to serve as a biomarker, a prespecified exploratory analysis yielded a strong association with PFS. Here, the addition of alisertib led to a marked clinical PFS benefit over paclitaxel alone in c-Myc–positive patients (HR, 0.29), whereas the opposite pattern was observed in the c-Myc–negative subgroup (HR, 11.8; Figure 1). A prospective study is needed to further validate the predictive value of c-Myc.
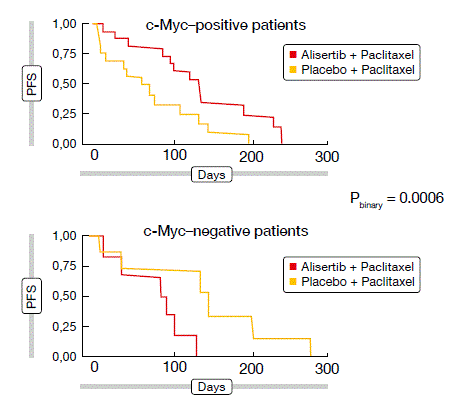
Figure 1: Effect on PFS of addition of alisertib to paclitaxel, according to c-Myc protein expression
Preliminary results with atezolizumab
The humanised monoclonal anti–PDL1 antibody atezolizumab has demonstrated promising clinical activity and a tolerable safety profile in a number of NSCLC clinical trials. As SCLC shows a high frequency of somatic mutations, these tumours might be amenable to treatment with PD-1/PD-L1 inhibitors. Sequist et al. presented the results obtained for an ES-SCLC cohort that was part of a larger phase Ia clinical trial investigating atezolizumab in locally advanced or metastatic solid tumours [6]. The first five patients were PD-L1–selected; after that, the enrolment continued regardless of PD-L1 expression status. Seventeen patients were evaluated in the safety and efficacy analyses.
Treatment with atezolizumab was generally well tolerated, with the majority of AEs as grades 1 or 2. Atezolizumab showed encouraging single-agent activity. The spider plot depicted in Figure 2 illustrates the responses achieved according to the immune-related response criteria (irRC). Objective responses per irRC occurred in 17.6 %. Disease control was obtained in 41.2 %. Median PFS was 2.9 months per irRC across all of the patients, and median OS was 5.9 months. In tumours expressing PD-L1, higher expression of PD-L1 mRNA and T-effector gene signature corresponded to a trend towards improved PFS (per irRC) and OS. The clinical benefit of atezolizumab continued beyond classical radiographic progression. A phase III randomised study of atezolizumab or placebo plus carboplatin/ etoposide in patients with ES-SCLC is currently recruiting (NCT02763579).
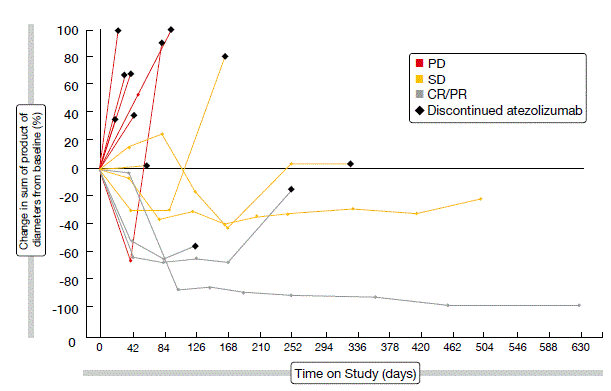
Figure 2: Confirmed responses to atezolizumab in patients with ES-SCLC (according to irRC)
Other novel agents
Chu et al. presented encouraging data on the monoclonal antibody BMS-986012 that targets fucosyl-GM1, which is a chemically defined monosialoganglioside that shows limited expression in normal tissues, but is highly expressed on the surface of tumour cells in SCLC [7]. BMS-986012 was developed as a first-in-class fully human immunoglobulin G1 monoclonal antibody. The anti-tumour activity of this agent is based on antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and antibody-dependent cellular phagocytosis.
The phase I/II dose-escalation and dose-expansion CA001-030 study was initiated in patients with relapsed and refractory SCLC. According to the preliminary data from the phase I monotherapy portion of this trial, BMS- 986012 was well tolerated, and patients responded to treatment. However, due to the small number of patients in this part of the study and their heterogeneity, no firm conclusions regarding the efficacy of BMS-986012 can be drawn yet. The BMS-986012 pharmacokinetics were consistent with what might be expected for a monoclonal antibody, and no anti-drug antibodies were detected in treated patients. Enrolment in the phase II monotherapy portion of this trial is ongoing. Further studies are currently investigating BMS-986012 as part of the combination regimens with nivolumab and chemotherapy.
In contrast, negative trial results were obtained with roniciclib, an oral, highly potent, small-molecule inhibitor of cyclin-dependent kinases [8]. A phase II, randomised, double-blind, placebo-controlled study conducted in patients with ES-SCLC compared cisplatin/ etoposide with carboplatin/ etoposide as first-line therapy in combination with roniciclib or placebo. No improvements were observed for the addition of roniciclib with regard to PFS, OS, ORR and time to progression. Moreover, the combination was not well tolerated, as patients who received roniciclib showed higher incidence of clinically important AEs and fatal AEs than those in the control group. The study was terminated following primary completion.
REFERENCES
- Alvarado-Luna G, Morales-Espinosa D, Treatment for small cell lung cancer, where are we now? -a review. Trans Lung Cancer Res 2016; 5: 26-38
- Ali S et al., Small cell lung carcinoma harbors gene fusions including MYCL1 fusions which can respond to aurora kinase inhibitors. ESMO 2016, abstract 14240
- Sos ML et al., A framework for identification of actionable cancer genome dependencies in small cell lung cancer. PNAS 2012; 109: 17034- 17039
- Hook et al., AACR 2010, abstract 2615
- Owonikoko TK et al., Randomized phase 2 study of the investigational aurora A kinase (AAK) inhibitor alisertinb (MLN8237) + paclitaxel versus placebo + paclitaxel as second-line therapy for small cell lung cancer (SCLC). ESMO 2016, abstract 14230
- Sequist LV et al., Clinical activity, safety and predictive biomarker results from a phase Ia atezolizumab trial in ES-SCLC. ESMO 2016, abstract 1425PD
- Chu Q et al., A phase 1/2 trial of a monoclonal antibody targeting fucosyl-GM1 in relapsed/refractory small cell lung cancer (SCLC): safety and preliminary efficacy. ESMO 2016, abstract 1427PD
- Reck M et al., Phase II study of roniciclib in combination with cisplatin/etoposide or carboplatin/ etoposide as first-line therapy in patients with extensive-disease small-cell lung cancer. ESMO 2016, abstract 1426PD