Diagnostics of EGFR-mutant disease: biomarkers with significant clinical implications
Alterations in multiple oncogenic pathways drive progression
The clinical relevance of additional genetic alterations in advanced EGFR-mutant NSCLC is not clear. Blakely et al. hypothesised that co-occurring genomic alterations in cancer-related genes can cooperate with the mutant EGFR to drive de-novo resistance to EGFR TKI treatments [1]. The investigators performed targeted exome sequencing of plasma cell-free DNA (cfDNA) in 86 samples collected from 81 patients with known clinical history. They found alterations in multiple concurrent oncogenic pathways, including TP53, WNT, PI3K and MYC, and in cell-cycle genes (e.g., CDK4/6, Cyclin D/E), which appeared to function collaboratively in tumour progression and drug resistance. Co-occurring aberrations were more frequently observed in EGFR-TKI nonresponders and they increased with each line of therapy. A proposed new model of EGFR-mutant NSCLC pathogenesis that arises from these findings suggests that TP53, RTK and RAS-MAPK are the most commonly coaltered functional genes. Alterations in cell-cycle genes showed the strongest correlations with non-response to EGFR TKI treatments, which warrants further investigation. As the authors noted, these findings call for re-evaluation of the prevailing paradigm of monogenicbased molecular stratification to monotherapy, and they highlight an alternative model of genetic collectives as a determinant of lung cancer progression and therapy resistance.
Predictive impact of early plasma clearance
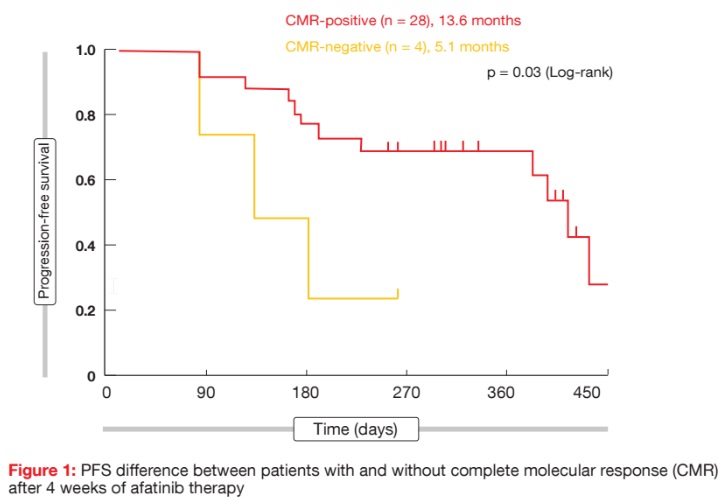
Liquid biopsy has been approved as an alternative method for the detection of clinically relevant EGFR mutations in NSCLC. Two analyses evaluated whether the early assessment of molecular responses in plasma can predict the clinical benefit of EGFR-targeted therapy. Otsubo et al. conducted a prospective, multi-institutional study of liquid biopsies with 57 patients who received afatinib monotherapy 40 mg/d [2]. Complete molecular response (CMR) was defined as mutant allele event/ frequency of exon 19 deletion or exon 21 L858R that was below the cut-off for positivity by digital PCR. Among the patients who were positive for EGFR mutation in plasma at baseline, 60.6 % and 87.5 % achieved CMR at 2 and 4 weeks, respectively. Patients with CMR at 2 weeks had longer PFS than those without (13.6 vs. 7.5 months; p = 0.11). There was a significant PFS benefit that favoured the group that obtained CMR at 4 weeks, compared to the population who did not (13.6 vs. 5.1 months; p = 0.03; Figure 1).
According to an exploratory analysis by Thress et al., the absence or presence of plasma EGFR mutations within 6 weeks of initiation of osimertinib therapy can be used to predict subsequent outcomes in patients with T790Mpositive advanced NSCLC [3]. Plasma samples from 143 patients who were participating in the phase I AURA trial were analysed for detectable EGFR mutations at baseline and 6 weeks after osimertinib treatment. The patients for whom both EGFR-sensitising and T790M mutations had disappeared at 6 weeks experienced significantly longer PFS than those with detectable mutations (10.8 vs. 4.2 months; p < 0.0001). This also applied to ORR (74 % vs. 41 %; p < 0.0001).
These results were also validated in an independent cohort of patients from the AURA2 and AURA3 studies who received osimertinib as second-line therapy. Again, patients with plasma EGFR mutation clearance at 6 weeks showed significant improvements with regard to both PFS (11.1 vs. 5.7 months; p = 0.001) and ORR (87 % vs. 53 %; p = 0.001). Further research will improve the understanding of whether continued detection of mutations at 6 weeks might indicate the presence of heterogeneous resistance mechanisms, which have the potential to be targeted by combination therapies.
EGFR T790M detection in exhaled breath condensate
The EGFR T790M somatic mutation is the most common mechanism of resistance to EGFR TKIs in NSCLC. As patients with advanced disease are not always amenable to repeated tissue biopsy for further molecular analysis, the development of minimally invasive methods to detect T790M mutation in cfDNA in the absence of tissue is being actively pursued. A pilot study explored the potential of exhaled breath condensate analysis as a novel method of T790M detection [4]. Exhaled breath condensate is an easily collected sample source and is known to contain cfDNA, including lung cancer mutations. Twenty-six patients were enrolled, who were either receiving first-generation or second-generation EGFR TKI therapy or had already developed T790M mutation before or during osimertinib treatment. Indeed, it was possible to detect the T790M mutation in the exhaled breath condensate using a commercially available targeted assay. These results suggest that exhaled breath condensate testing is responsive to recognised dynamic molecular changes that occur on TKI treatment. The authors suggested that exhaled breath condensate analysis might be an attractive alternative for future research to optimise the detection of the T790M mutation in liquid biopsies.
Resistance to third-generation TKIs: osimertinib …
Acquired EGFR C797S/G mutations have been identified as a major resistance mechanism to the third-generation EGFR TKI osimertinib, but other mechanisms relating to osimertinib are still largely unknown. Zhou et al. therefore conducted targeted nextgeneration- sequencing-based mutational profiling and in-vitro testing for osimertinib resistance mutations in 93 patients [5]. Most of these had adenocarcinoma and stage-IV disease. Twenty-nine percent of osimertinib resistant tumours showed secondary mutations on the C797, L792 or L718 residues of EGFR. The in-vitro data demonstrated that the L792 and L718 mutations induce resistance to osimertinib. Thus, these mutations represent alternative resistance mechanisms in addition to the wellknown C797S mutation. The authors noted that in patients with C797, L792 and L718 wild-type, MET and KRAS copy number gains might serve as bypass resistance mechanisms.
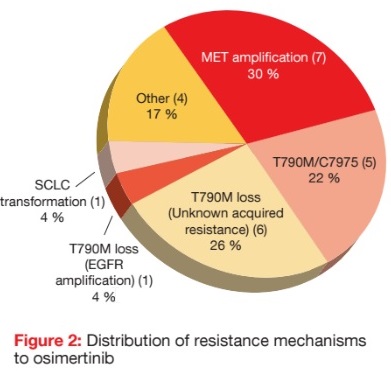
Indeed, as demonstrated by a retrospective analysis of 23 patients from the phase I AURA trial, MET amplifications represent a major resistance mechanism to osimertinib treatment [6]. All patients underwent tissue biopsy and/ or analysis for plasma circulating tumour DNA at the time of progression on osimertinib. None of them had shown MET amplification prior to the start of osimertinib therapy. According to this analysis, all of the patients retained the initial EGFR mutation on postosimertinib testing. MET amplification was observed in 30 % of cases by tissue or plasma analysis, and was the most common resistance mechanism in this cohort (Figure 2). EGFR T790M/C797S mutation emerged in 22 % of cases. T790M ‘loss’ was also commonly seen (26 %), typically with no identified resistance mechanism. The authors noted that patients with MET amplification responded to subsequent therapies containing MET inhibitors. Clinical trials testing both MET and EGFR inhibitors are ongoing. However, a substantial minority of patients in this study had no identifiable drivers of resistance to osimertinib, which calls for further research efforts in this area.
… and nazartinib
The third-generation EGFR TKI nazartinib is highly effective against EGFR-activating mutations and T790M resistance mutations. A phase I dose-escalation trial yielded favourable results, with an ORR of 47 %, DCR of 87 %, and a median PFS of 9.7 months [7]. However, resistance invariably arose. The analysis conducted by Tan et al. assessed the genetic characteristics of tumour biopsies obtained at baseline and upon tumour progression from patients treated with nazartinib in the phase I/II study, to identify mechanisms of resistance [8]. The data presented at the ASCO Congress were from the phase I dose-escalation part.
At baseline, the most frequent genetic alterations included TP53 mutations and loss of CDKN2A/2B. No relationship was observed between the allelic frequency fraction of T790M and the depth or duration of response. There were no correlations between any genetic alteration and PFS. After progression, T790M was not detectable in five out of eight cases (62.5 %) in tumours that had been T790M-positive at baseline, which suggests that nazartinib is a highly active inhibitor of T790M-positive clones. In this population, however, concomitant mutations were detected, including potential resistance alterations, such as EGFR G724S mutation, MET amplification, BRAF fusions, and mTOR deletion alteration. Thirty-seven percent of patients remained T790M-positive, with one individual acquiring EGFR C797S mutation. These genetic alterations provide hypotheses to guide future combination treatments with nazartinib to prevent or delay the emergence of resistance.
References:
- Blakely CM et al., Evolution and clinical impact of genomic alterations detectable in circulating tumor DNA of 1150 EGFR-mutant lung cancer patients. ASCO 2017, abstract 9009
- Otsubo K et al., Predictive impact of complete molecular response in plasma: A phase II, liquid biopsy study in EGFR mutated NSCLC patients treated with afatinib (WJOG 8114LTR). ASCO 2017, abstract 9027
- Thress KS et al., Clearance of plasma EGFR mutations as a predictor of outcome on osimertinib in the AURA trial. ASCO 2017, abstract 9018
- Smyth R et al., The novel detection of EGFRT790M mutations in exhaled breath condensate. ASCO 2017, abstract 9032
- Zhou C et al., Investigating novel resistance mechanisms to third generation EGFR TKI osimertinib in non-small cell lung cancer patients using next generation sequencing. ASCO 2017, abstract 2572
- Piotrowska Z et al., MET amplification (amp) is a major resistance mechanism to osimertinib. ASCO 2017, abstract 9020
- Tan S-W et al., Updated results of a phase 1 study of EGF816, a third-generation, mutantselective EGFR tyrosine kinase inhibitor (TKI), in advanced non-small cell lung cancer (NSCLC) harboring T790M. J Clin Oncol 34, 2016 (suppl; abstr 9044)
- Tan S-W et al., Genomic profiling of resistant tumor samples following progression on nazartinib (EGF816), a third generation, mutantselective EGFR tyrosine kinase inhibitor (TKI), in advanced non-small cell lung cancer (NSCLC). ASCO 2017, abstract 11506
More posts
Anti-angiogenic and immunotherapeutic approaches in mesothelioma
Malignant pleural mesothelioma (MPM) is a rare tumour that is often diagnosed at an advanced stage. Limited efficacy of the available therapies contributes to the generally poor prognosis for MPM patients. Since 2003, the only approved regimen for MPM treatment has been chemotherapy with pemetrexed and cisplatin, with median survival of approximately 12 months.
Interview: Lung cancer in China: hurdles and progress
Lung cancer is a considerable issue in China. Every year, we have 700,000 new cases. There is a need to perform clinical trials and to launch innovative drugs. With regard to the introduction of targeted therapies, China lags 3 to 4 years behind when compared to the western countries. Two months ago, the EGFR TKI afatinib was launched, offering Chinese patients with EGFR-mutant lung cancer an effective treatment option.
Real-world utility of ctDNA NGS to identify matched targeted therapy
Liquid biopsy for plasma circulating tumour DNA (ctDNA) next generation sequencing (NGS) is a rapidly evolving science. Plasma ctDNA assays are now commercially available, and are increasingly adopted in the community with a paucity of evidence-based guidance on timing and value of this test. Sabari et al. sought to determine the feasibility and utility of plasma ctDNA NGS to identify matched targeted therapy in a real-world clinical setting.
Further defining the optimal use of immune checkpoint inhibitors
As the anti-PD-1 antibody nivolumab is known to induce deep and durable responses in a subset of lung cancer patients, this agent was investigated in the neoadjuvant setting, which is an area of unmet need. There have been no advances in systemic treatment of resectable lung cancer since 2004. Chaft et al. hypothesised that neoadjuvant nivolumab treatment might induce immunity against micrometastases.
Established targeted agents taking root in the HER2-positive setting
HER2 aberrations in lung cancer are being increasingly identified due to the use of sensitive testing procedures, such as multiplexed testing and next-generation sequencing. Mutations of the HER2 gene need to be distinguished from HER2 amplifications and HER2 protein overexpression. In contrast to breast and gastric cancer, HER2 overexpression in NSCLC does not always occur with HER2 amplification, while amplifications and HER2 mutations are generally mutually exclusive.
Diagnostics of EGFR-mutant disease: biomarkers with significant clinical implications
The clinical relevance of additional genetic alterations in advanced EGFR-mutant NSCLC is not clear. Blakely et al. hypothesised that co-occurring genomic alterations in cancer-related genes can cooperate with the mutant EGFR to drive de-novo resistance to EGFR TKI treatments. The investigators performed targeted exome sequencing of plasma cell-free DNA (cfDNA) in 86 samples collected from 81 patients with known clinical history.