

Biomarker-based clinical trials: study design and regulatory requirements
Introduction
Due to the recent developments in the field of molecular diagnosis, it has become evident that there is no such entity as cancer per se, as there are considerable biological differences even within a particular anatomical subtype. Genomic characteristics do not only help to better define tumor subtypes, but frequently they provide the opportunity to target these subtypes as well. This can thus lead to the development of therapeutic approaches that surpass conventional treatments in terms of efficacy and tolerability. The International Cancer Genome Consortium (http://icgc.org/) has made it its task to generate comprehensive catalogues of genomic abnormalities (somatic mutations, abnormal gene expression, epigenetic modifications) in tumors from 50 different cancer types and/or subtypes across the globe, and to make these data publicly available. This is one of the largest natural history projects of the last decades.
The availability of rapid and relatively cheap molecular profiling underpins translational research, enabling the transition from preclinical insights to global implementation of testing of new treatments in clinical trials. The inclusion of companion biomarkers has become mandatory in all new drug trials that are conducted for targeted therapies in the oncological field. Whilst the term ‘biomarker’ encompasses a broad range of parameters that go beyond the molecular characteristics, it has become most closely associated with genomics and other ‘-omic’ analyses.
Definition
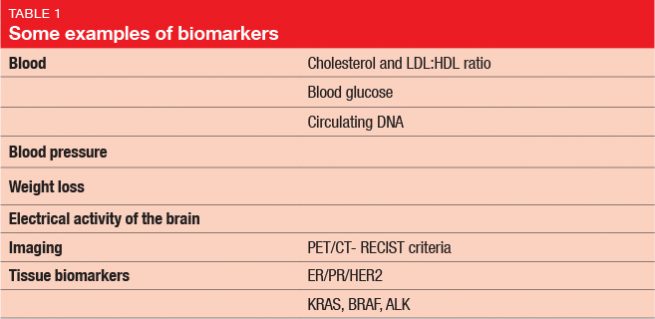
In 1998, the National Institutes of Health Biomarkers Definitions Working Group defined a biomarker as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention”. This means that biomarkers represent a variety of parameters including, but not limited to, the genomic characteristics. Table 1 provides some examples of biomarkers. Even the diagnosis itself, or the histology of the tumor, count as biomarkers, as well as patient age and patient sex. Drug metabolism is an important aspect too, although this is often overlooked. With respect to pharmacokinetics, potential biological markers include drug absorption, metabolism, distribution, and excretion. The differences between patients and their ability to break down drugs (i. e., fast or slow metabolizers) can be crucial for bioavailability and toxicity. Pharmacogenomics, which evaluates the role of a patient’s genetic set-up in their responses to treatments, has risen to great importance. Genomic markers are targeted directly by certain therapies, and can influence their effectiveness.
Prognostic and predictive biomarkers
While prognostic biomarkers hint at the likely outcome of a patient with a particular disease regardless of their treatment, predictive biomarkers provide information on whether that patient will respond to a certain agent. Some markers provide both of these functions. For instance, estrogen receptor positivity in breast cancer patients indicates a favorable prognosis (providing prognostic information); at the same time, it means that the patient will probably respond to endocrine therapy (providing predictive information).
Surrogate biomarkers
In early phase trials, surrogate biomarker endpoints can allow the determination of treatment effects at earlier times than the ultimate clinical endpoint of interest, such as progression-free survival or overall survival. Valid surrogate biomarkers have both prognostic and predictive properties, as they also demonstrate mechanisms of molecular action. However, the question remains whether the treatment effect on the biomarker reliably predicts the treatment effect on the clinical endpoint.

Use of biomarkers in trials
In clinical trials, biomarkers serve a range of practical uses:
- Screening of patients for eligibility
- Stratification into subgroups
- Monitoring of responses
- Correlative studies for future reference
Many study protocols include the collection of additional samples that are not necessarily used for the endpoint of the specific trial, but rather for purposes of design of future studies. Also, studies can be designed with a view to re-analyzing the dataset later on in the light of new information that has been revealed by further research (e. g., new markers or genes).
Ideal markers require minimally invasive sampling, are reliable (i. e., internally consistent), cheap, and highly sensitive and specific. Re-biopsy is frequently recommended in the advanced setting, as clonal evolution can lead to differences in molecular characteristics between the original tumor and its metastases, and even between metastatic lesions within a given patient. Among other uses, biomarkers can serve as indicators of futility that alert the researchers to the necessity of early study termination; for instance, if the biological effect is below the maximally tolerated dose.
Basket and umbrella trials
The emergence of genomic biomarkers has prompted the development of trials that are not restricted to certain anatomically defined cancer types. In these so-called ‘basket trials’, a particular mutation is targeted that occurs in tumors of different origins. ‘Umbrella trials’, on the other hand, assess a variety of drugs usually in one cancer type that has different molecular alterations. Here, patients with a single tumor type or histology are enrolled, but multiple sub-trials evaluate targeted therapies within molecularly defined subsets. The implementation of basket trials and umbrella trials poses specific challenges both in terms of recruitment, since the numbers of eligible patients falls with increasing use of molecular inclusion criteria, and also with needing to create trials that require cooperation from pharmaceutical drug companies that might have competing agents in their pipeline.

Ethical and practical limitations
From an ethical point of view, it could be argued that molecular screening should be performed at the time of diagnosis rather than in the advanced disease stage, as this can enable patients to benefit from certain treatments early on. However, early testing raises financial concerns if the patient has to pay for this themselves, but also in terms of cost-benefit evaluations if the government pays. Another ethical conflict that might arise is the randomization of patients into the control group. Many patients decline this, in the knowledge that they might be denied an enormously effective treatment.
Practical problems also arise because biomarkers tend to be imprecise. More widespread genomic analysis shows that the penetrance of genes is often incomplete, and some major confounders are awaiting improved understanding, such as the microenvironment. For example, the bacteria in the gut are known to affect the way patients respond to treatments. Also, further research is required regarding putative interactions between the targeted pathways and the immune system. Moreover, objective measurability, as mentioned in the “Definition” section above, can pose problems in clinical practice. While the presence of mutations is assessed on a yes-or-no basis, immunohistochemistry uses ranges of values that often elude precise quantification.
It can be assumed that data from phase IV registries will accumulate in the future and will shed light on these issues. Pharmaceutical companies are obliged to set-up these registries, which correlate the presence of biomarkers to clinical outcomes.

Validation of prognostic and predictive biomarkers
From the registration point of view, biomarkers need to be valid and reproducible. Regulatory authorities are required to ensure that standardized tests are available for biomarker testing. The average pathology laboratory should be able to deliver them in a rapid, cheap, and effective manner. Equally important for registration, the biomarker needs to be included in any reimbursement schedule.
Validation of biomarkers in clinical trials includes:
- Proof of concept;
- Experimental validation;
- Analytical performance validation;
- Protocol standardization.
Proof of concept
A classic example of proof-of-concept assessment is the identification of the KRAS mutational status as a predictive marker in the context of anti-EGFR antibody therapy. KRAS mutation has been established as an important marker in colorectal cancer (CRC) since the 1990s. The KRAS protein regulates downstream proteins in the EGFR signaling pathway that are associated with tumor survival, angiogenesis, proliferation, and metastasis. Whereas wild-type KRAS denotes preserved function of the protein, mutant KRAS is indicative of persistent growth stimulation. This mutation is found in one third of CRC patients.
The primary analysis of the randomized, phase III NCIC/AGITG CO.17 trial, which did not stratify for KRAS mutation status, yielded no clinically relevant advantage of the anti-EGFR antibody cetuximab over placebo in patients with advanced CRC [1]. According to the post-hoc analysis, however, when mutations status was considered, there was a distinct difference between the two treatment arms [2]. In KRAS-mutant patients, cetuximab did not show any benefit, whereas those with KRAS wildtype obtained significant overall survival advantage when treated with this antibody.
Experimental validation
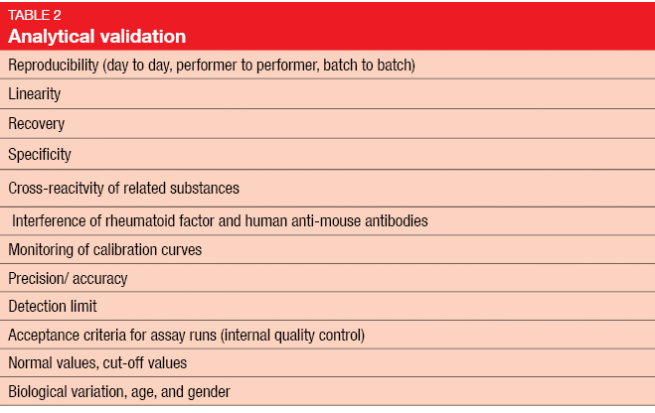
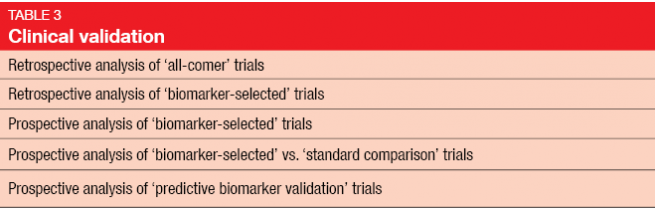
Once biomarker development reaches the level of the diagnostic laboratory, very high standards are required. Experimental validation includes the two elements of analytical and clinical validation (Tables 2 and 3) [3]. Clinical validation should ideally be conducted in a prospective study, rather than in the retrospective setting, as biomarker analysis on the basis of existing data from randomized controlled trials has several drawbacks [4]. In many cases, the original study will not have been powered for a correlative science endpoint, and tissue has not necessarily been obtained in all of the randomized patients. Outcomes obtained in a retrospective setting are generally considered hypothesis-generating and need to be confirmed in prospective studies.
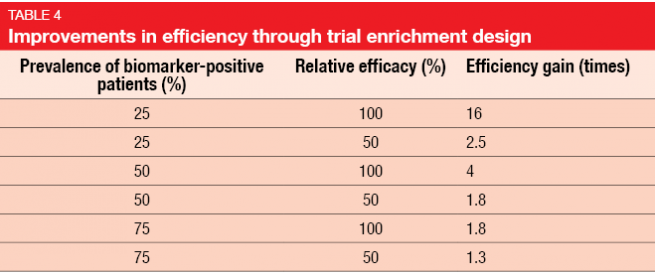
Gains in efficiency depend on the marker prevalence and the relative efficacy in biomarker-positive and biomarker-negative patients. Trials with an enrichment design only enroll patients who are likely to respond. The use of an enrichment design improves efficiency (Table 4) and is also an important factor with regard to cost [5]. However, exclusion of those patients who are not likely to benefit implies the need to understand the scientific basis of the targeted agent, and statistical simulations should have demonstrated improved efficacy through enrichment.
Analytical performance validation
Analytical performance validation encompasses clinical laboratory measurements with the aim of assessing the analytical performance of various biomarker assays. For KRAS mutation testing, seven different methodologies were compared in 2009 [6]. Today, next-generation sequencing is increasingly becoming the standard technique as part of a much broader panel to examine mutations across several targetable pathways.
Protocol standardization
With respect to KRAS testing, data from the CRYSTAL study suggested that extended RAS testing is more appropriate, as mutations in various KRAS exons as well as in NRAS exons equally affect outcomes [7]. Today, all-RAS mutation testing is recommended by the guidelines [8].

Approval of biomarker studies: regulatory requirements
Regulatory authorities are generally perceived as being difficult and bureaucratic, but they serve an important function in terms of protecting and promoting public health. They ensure quality, safety, and efficacy of treatment. Also, they provide adequate and appropriate information for both patients and physicians. Naturally, their approach is conservative and careful, as mistakes on their part can have vast consequences. From the researcher’s point of view, being empathetic to their objectives and complying with them will alleviate cooperation and reduce conflict. The instructions of the regulatory authorities should thus be followed closely.
The approaches to regulation vary worldwide. For instance, the Japanese regulatory authorities believe that clinical data from foreign patients are of limited applicability to Japanese patients, which is why they require stand-alone development programs. However, there are many similarities. Regulatory authorities generally run comprehensive websites that provide useful information on biomarker qualification, such as numerous academic inputs into the optimal study design. Templates where information can be added in to a prepared structure are provided at the US Food and Drug Administration (FDA) website (www.fda.gov). At the European Medicines Agency (EMA) website (http://www.ema.europa.eu/ema/), researchers have the opportunity to receive comments from member states as part of the centralized approval procedure (Figure 1).
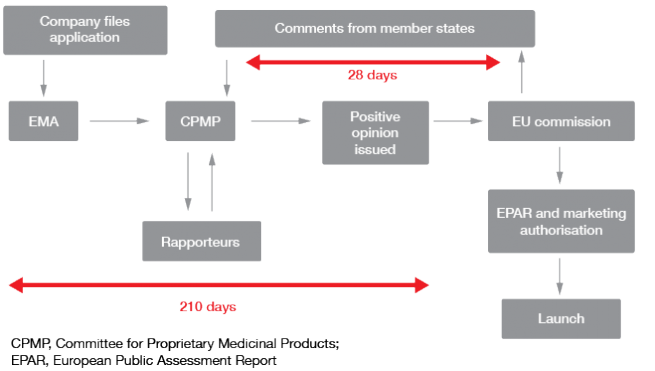
Figure 1: EMA: centralized approval procedure
Review and protocol
The regulatory authorities are still assessing the optimal ways to incorporate biomarkers into clinical trials. For the future, it can be expected that countries will increasingly cooperate in this respect.
At present, researchers have to provide protocols that detail all of the proceedings planned for the study, as well as the quality assurance measures. In most cases, the rationale for a trial will be supported by experimental findings provided by other study groups. Only a minority of researchers can provide their own laboratory data. Therefore, a detailed review of the available literature is necessary as a background statement. The FDA requires researchers to create a Context of Use Statement for Biomarker Qualification, which is part of the general protocol and describes the manner of use, interpretation, and purpose of use of a biomarker in drug development [9]. Five elements of the Context of Use Statement are specifically defined (Table 5). In the context of quality improvement and quality assurance, even apparently small aspects can be of importance, such as correct handling of blood tubes or timing of assessments by a pathologist. All of these should be defined in the study protocol.

Translational research in NSCLC
The transformation of the management of advanced non–small-cell lung cancer (NSCLC) over the last 10 years exemplifies the magnitude of changes in treatment paradigms that can be brought about by scientific progress. Translational research has contributed greatly to each step along the way.
Enzyme expression as a key to efficacy
Until 2008, the choice of treatment of lung cancer patients relied mainly on differentiation between small-cell and non–small-cell histology, with doublet chemotherapy being the standard approach. Toxicity was an important determinant in any treatment decision.
At that time, the first prospective trial was conducted in NSCLC patients that contained a pre-planned analysis to evaluate histology as a possible predictive factor [10]. This showed that pemetrexed plus cisplatin is more efficacious in patients with adenocarcinoma, while the results obtained in patients with squamous-cell histology favored gemcitabine plus cisplatin. These findings prompted a revision in the indications for pemetrexed (which is now approved for the treatment of advanced, non-squamous NSCLC) and underlined the relevance of this multidisciplinary approach to disease.
Translational research provided the rationale for the then-unusual histological stratification. It was shown that thymidylate synthase (TS), which is one of the enzymes involved in pemetrexed metabolism, is expressed to a significantly higher degree in squamous-cell carcinoma than in adenocarcinoma of the lung (Figure 2) [11]. Meanwhile, a meta-analysis confirmed the predictive significance of TS expression for several endpoints (i.e., response rate, progression-free survival, overall survival) in the setting of pemetrexed-based chemotherapy in NSCLC [12].
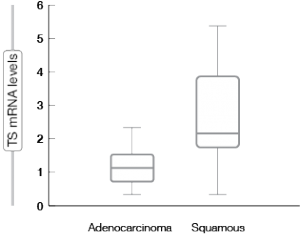
Figure 2: Significant differences in the expression of thymidylate synthase (TS) in adenocarcinoma and squamous-cell carcinoma of the lung (formalin-fixed paraffin-embedded tissue)
For small-cell lung cancer (SCLC), on the other hand, the randomized GALES study revealed inferior performance of pemetrexed plus carboplatin as compared to the standard regimen of etoposide plus carboplatin [13]. These findings are in keeping with insights gained concurrently in the preclinical setting, according to which TS is highly expressed in SCLC [14].
Reasons for negative trial results
However, pharmacogenomic and translational research is not always successful. A testament to this is the adjuvant study landscape for NSCLC, which has been marked by a series of negative trials. Translational research can be conducted most easily in the adjuvant setting, due to the abundance of available tissue. There are several possible reasons for this. One is the diagnostic test itself, which can be tainted by insufficient sensitivity or specificity. The TASTE trial determined the expression of the excision repair cross-complementation group 1 (ERCC1) protein using this as the biomarker in the design of the pharmacogenomics-driven trial. However, the study was stopped at 150 patients due to the unexpected lack of reliability of the ERCC1 IHC read-out, and the phase III trial was canceled. Using historical International Adjuvant Lung Cancer Trial (IALT) data, the expected and observed biomarker distribution deviated considerably from each other [15]. The choice of detection and enrichment was presumably the reason for the negative outcome in the RADIANT trial, too. The RADIANT trial compared erlotinib and placebo following complete tumor resection and standard treatment [16]. Here, the researchers used IHC and FISH to determine EGFR status, which was probably not an adequate technique, even though it was considered modern at the time of the design of the study.
Another reason behind negative results in translational research is the choice of only one biomarker. As a general rule, a more complete profile needs to be defined in pharmacogenomic-driven trials. A single biomarker suffices only if it is a distinct driver aberration, and the EGFR mutation is a good example in this context. Generally speaking, all the pharmacogenomic-driven trials conducted with only one biomarker have been negative, in both the adjuvant and the advanced settings.
Trial design can also be flawed, which makes relevant comparisons difficult. Above all, the molecular profile should be defined prior to the allocation (or randomization) of the patients to the experimental arm versus the control arm, as used in the design of the adjuvant ITACA trial [17]. ITACA compared standard chemotherapy versus pharmacogenomic-guided regimens after the assessment of ERCC1 and TS using real-time PCR, with the identification of four different profiles.

REFERENCES
- Jonker D et al., Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357: 2040-2048
- Karapetis CS et al., K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359: 1757-1765
- Gandara et al., CAPR Workshop, April 2011
- Lee CK et al., Molecular biomarkers to individualise treatment: assessing the evidence. Med J Aust 2009; 190: 631-636
- Simon R, Maitournam A, Evaluating the efficiency of targeted designs for randomized clinical trials. Clin Cancer Res 2004; 10(20): 6759-6763
- Whitehall V et al., A multicenter blinded study to evaluate KRAS mutation testing methodologies in the clinical setting. J Mol Diagn 2009; 11(6): 543-552
- Van Cutsem E et al., Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 2015; 33(7): 692-700
- Van Cutsem E et al., ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016; 27: 1386-1422
- http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/ucm284620.htm
- Scagliotti GV et al., Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non–small-cell lung cancer. J Clin Oncol 2008; 26: 3543-3551
- Ceppi P et al., Squamous cell carcinoma of the lung compared with other histotypes shows higher messenger RNA and protein levels for thymidylate synthase. Cancer 2006; 107(7): 1589-1596
- Liu Y et al., Expression of thymidylate synthase predicts clinical outcomes of pemetrexed-containing chemotherapy for non–small-cell lung cancer: a systemic review and meta-analysis. Cancer Chemother Pharmacol 2013; 72: 1125-1132
- Socinski MA et al., Phase III study of pemetrexed plus carboplatin compared with etoposide plus carboplatin in chemotherapy-naive patients with extensive-stage small-cell lung cancer. J Clin Oncol 2009; 27(28): 4787-4792
- Monica V et al., Differential thymidylate synthase expression in different variants of large-cell carcinoma of the lung. Clin Cancer Res 2009; 15(24): 7547-7552
- Soria JC et al., Results of the prospective, randomized, and customized NSCLC adjuvant phase II trial (IFCT-0801, TASTE trial) from the French Collaborative Intergroup. J Clin Oncol 31, 2013 (suppl; abstr 7505)
- Kelly K et al., A randomized, double-blind phase 3 trial of adjuvant erlotinib (E) versus placebo (P) following complete tumor resection with or without adjuvant chemotherapy in patients (pts) with stage IB-IIIA EGFR positive (IHC/FISH) non–small-cell lung cancer (NSCLC): RADIANT results. J Clin Oncol 32:5s, 2014 (suppl; abstr 7501)
- Novello S et al., Preliminary results of the international tailored chemotherapy adjuvant trial: the ITACA trial. J Thorac Oncol 2015; 10(9), ORAL04.03