Novel early clinical approaches in solid tumors
CLDN6 CAR-T cell therapy – Encouraging results
Chimeric antigen receptor (CAR)-T cells have proven to be effective in the clinic for patients with malignant B-cell tumors but their application for solid tumors is challenging [1]. BNT211 is a novel therapeutic approach which comprises two components: CAR-T cells targeting the Claudin 6 (CLDN6) and a CLDN6-encoding CAR-T cell amplifying RNA vaccine (CARVac) [2]. On June 23, 2022, BNT211 has received priority medicine (PRIME) designation by the European Medicines Agency (EMA) for the third- or later-line treatment of testicular germ cell tumors [3]. The PRIME status was based on positive preliminary data from the ongoing BNT211-01 phase I/II study (NCT04503278) [4]. Updated data from this study were presented at ESMO 2022 [2].
The trial followed a 3+3 dose escalation design with bifurcation (Figure 1). Patients with an ECOG performance status of 0 or 1, CLDN6-positive tumors (≥ 50 % of tumor cells CLDN6-high [II/III+]) and no other treatment option were eligible. In part I of the phase I dose escalation study, patients received a dose of 1×107 CLDN6 CAR-T cells (dose level [DL] 1; n = 3) or 1×108 CAR-T cells (DL 2, n = 6) as monotherapy. In part two of the phase I trial, four patients received the DL 1 of the CAR-T cells plus CARVac and nine patients received DL 2 plus CARVac. Apheresis and CAR-T cell manufacturing were performed at Day -18 or earlier. Patients underwent lymphodepletion on Day -5 to Day -3. CAR-T cell infusion was done on Day 1 and dose-limiting toxicity (DLT) was then assessed for 28 days. Patients who also received the vaccine were administered the first dose on Day 4; the first five CARVac treatments occurred every three weeks (Q3W) according to a one intra-patient dose escalation schemata, followed by a treatment every six weeks thereafter. Of the nine patients who received DL 2 plus CARVac, one patient had 50 % lymphodepletion and two patients had no lymphodepletion.
The updated data read-out (data cut-off dates: June 15, 2022, for safety and August 16, 2022, for efficacy) included 22 patients (21 evaluable for efficacy) with a median age of 46 years, most patients being male (n = 15). This heavily pretreated population received a median of four (range, 3-9) prior lines of therapy. All patients showed a robust dose-dependent CAR-T cell expansion. A long-term persistence of CAR-T cells was detected for more than 100 days post CAR-T cell infusion, with a few patients showing a persistence for ≥ 200 days. Efficacy assessment showed a best overall response rate (ORR) of 33 % (7/21) and a disease control rate (DCR) of 67 % (14/21), with one patient having a complete response (CR), six patients with a partial response (PR) and seven patients with a stable disease (SD). Testicular cancer patients showed particularly encouraging responses at DL 2 (ORR 57 %, DCR 85 %; n = 7).
DLTs were observed in two patients, including one treated at DL2 monotherapy (prolonged pancytopenia after lymphodepletion) and one at DL2 plus CARVac (hemophagocytic lymphohistiocytosis before start of CARVac). A total of 13 patients experienced treatment-emergent adverse effects (TEAEs) of grade 3 or higher suspected to be related to the investigational agents. These TRAEs consisted mainly of lymphodepletion or asymptomatic elevations of lipase and transaminases. Cytokine release syndrome (CRS) was observed in ten patients (45 %) and associated with high interleukin-6 (IL-6) levels. All instances of CRS were grade 3 or less and manageable with tocilizumab, if needed. The only reported case of grade 1 immune effector cell-associated neurotoxicity syndrome quickly resolved. A total of eight patients died due to disease progression.
The new dataset from the BNT211-01 study underscored the promising previously reported results obtained with the combination of CLDN6 CAR-T cells and CARVac. Moreover, the process of CLDN6 CAR T-cell generation was switched to an automated process and the recommended phase II dose escalation is still ongoing.
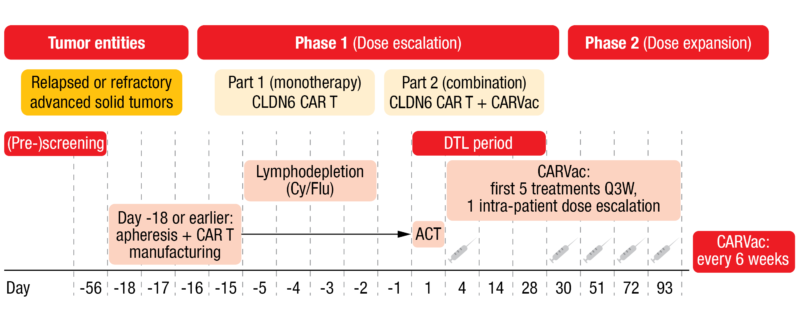
Figure 1: BNT211-01 trial design
GDFATHER-1 trial of visugromab
Growth and differentiation factor 15 (GDF-15), a TGF-β superfamily member, has been associated not only with anorexia but more importantly also with potent local immunosuppression under physiological and pathophysiological conditions [5]. GDF-15 is overexpressed by several solid tumors, associated with the resistance to anti-PD1/PD-L1 treatments and high levels have been linked to a limited survival of the affected patients. Once secreted by the tumor, GDF-15 prevents T-cell migration into the tumor and suppresses T-cell function as well as the adaptive immune response in the tumor microenvironment [6, 7]. The first-in-human trial GDFATHER-1 (NCT04725474) investigated the GDF-15 neutralizing antibody visugromab – formerly known as CTL-002 – combined to an anti-PD1 immune checkpoint inhibitor (ICI) in patients with relapsed or refractory tumors; the final results were presented at this year’s ESMO meeting [7].
Eligible patients with an advanced-stage mixed solid tumor in last-line treatment (including those relapsed/refractory to prior anti-PD1/PD-L1 therapy) received escalating doses of visugromab intravenously (IV) (0.3-20 mg/kg, Q2W) in this 3+3 phase I trial in a “monotherapy-followed-by-combination”- design: visugromab was first given as monotherapy and thereafter combined with the anti-PD-1 ICI nivolumab. Biopsies were taken consecutively at Day 0, 14 (monotherapy) and 28 (combination). The analysis of the tumor material showed increased CD4+ and CD8+ T-cells in most of the patients.
At enrollment, patients already received a median of 4.3 prior lines of treatment. The number of Ki67+ and GranzymeB (GrzB)+ T-cells was increased 2-fold in 55 % and 50 % of evaluable patients, respectively, demonstrating a proof-of-mechanism of this study design. Regarding clinical activity, the ORR according to RECIST v1.1 reached 17 % for DL 3-5, with six of 18 patients (33.3 %) experiencing a significant clinical benefit. Three of those patients (50 %), who received up to six lines of prior therapy, achieved a confirmed PR with up to twelve months duration (one patient was still on treatment at the time of analysis). The other three patients had a long-term disease stabilization (SD), with one moving to a PR after local irradiation of a single progressive lesion. The tumor regression rate for DL 3-5 and DL 4-5 were 22 % and 25 %, respectively. Additionally, two potential predictive biomarkers were identified and will be further investigated in a biomarker-selected cohort.
Regarding safety, the combination of visugromab and nivolumab showed an excellent tolerability, with no DLT and grade ≥4 AEs. The pharmacodynamic analysis confirmed the complete neutralization of GDF-15.
In conclusion, visugromab monotherapy followed by a combination with nivolumab has demonstrated a clinically meaningful tumor response, as well as an excellent safety and tolerability in last-line anti-PD1/PD-L1 relapsed/refractory solid tumor patients. To validate those primarily encouraging results, visugromab is currently in phase II development with signal-finding studies in five indications, a predictive biomarker-selected cohort and in the neoadjuvant setting.
BI 907828: an MDM2-p53 antagonist
The tumor suppressor protein p53 is activated upon various stress signals and mediates downstream cellular responses, including amongst others cell-cycle arrest, DNA repair, senescence, and apoptosis [8]. MDM2 is a negative regulator of p53; of note, the auto-regulatory feedback loop between MDM2 and p53 is essential to keep a low p53 level and to limit aberrant p53 activity [8, 9]. Approximately 5-7 % of tumors present with MDM2 amplifications [10]. By binding to free MDM2, the BI 907828 acts as a highly potent, orally available antagonist of the interaction between MDM2 and p53 thereby restoring p53 function [11] . Preclinical data in TP53 wild-type, MDM2-amplified dedifferentiated liposarcoma (DDLPS) patient derived xenograft models have already shown the antitumoral activity of BI907828 [11]. Preliminary data of the ongoing phase Ib (dose expansion) study (NCT03449381) were presented at ESMO 2022 [12].
Currently, patients with TP53 wildtype (wt), MDM2-amplified advanced solid tumors are recruited to cohort I (sarcomas, any line) or cohort II (NSCLC, gastric-, urothelial-, pancreatic- and biliary tract-cancers, 2nd and later lines). As of July 2022, 107 patients have received BI907828 orally across all dose levels; among them, 39 patients had DDLPS, 16 patients had well-differentiated liposarcoma (WDLPS), and four patients had biliary tract cancer. These heavily pretreated patients (median of 2 prior treatment lines; range, 0-11) had a median age of 57 years, were predominantly male (55.1 %), Caucasian (70.1 %) and had a very good performance status (ECOG 0, 58.9 %).
The maximum tolerated dose of BI 907828 was 60 mg Q3W; the recommended dose for expansion was selected as 45 mg, Q3W. BI907828 showed a long half-life of around 30 to 60 hours; a linear correlation between the target engagement biomarker GDF-15 and patient exposure was detected, too (70-fold increase of GDF-15 over baseline). Among the 94 evaluable patients included in the efficacy analysis, 79 achieved at least a stable disease (DCR, 84.0 %). In total, twelve patients had a confirmed PR, most of them were MDM2-amplified (ORR, 12.8 %) (Figure 2). Overall, all 16 WDLPS-patients achieved at least a stable disease (DCR, 100 %), with nine patients receiving study treatment for at least nine months. Of the 36 DDLPS-patients, 32 achieved at least stable disease (DCR, 88.9 %) and the preliminary median PFS was 8.1 % (13 patients are still on treatment).
Nausea (in 74.1 % of patients) was the most common treatment-related adverse event (TRAE) any grade, thus, antiemetic prophylaxis or treatment was implemented. The most frequent grade ≥3 TRAEs experienced by patients receiving the recommended dose of 45 mg were neutropenia (20.3 %), thrombocytopenia (18.6 %) and anemia (10.2 %).
In this phase Ib study, the MDM2-p53 antagonist BI907828 demonstrated a manageable safety profile and encouraging preliminary antitumor activity in patients with solid tumors. The efficacy was particularly promising in patients with MDM2-amplified DDLPS, WDLPS, and biliary tract cancer. An ongoing phase II/III study (Brightline-1, NCT05218499) is currently investigating the safety and efficacy of BI 907828 compared to doxorubicin as first-line treatment for patients with advanced DDLPS.
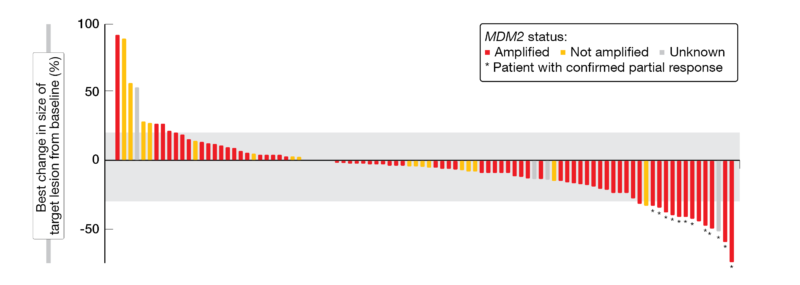
Figure 2: Waterfall-plot indicating the best change in size of target lesions from baseline
Antitumor activity of B7-H3 DXd antibody-drug conjugate
The B7 homologue 3 (B7-H3) – a transmembrane protein belonging to the B7 family – is frequently overexpressed in many tumors and associated with a poor prognosis [13-15]. DS-7300 is an antibody-drug conjugate (ADC) comprising a humanized anti-B7-H3 IgG1 monoclonal antibody conjugated to a topoisomerase I inhibitor payload via a tetrapeptide-based cleavable linker [16]. Part I of the phase I/II DS7300-A-J101 study (NCT04145622) previously showed that DS-7300 was well tolerated with early signs of antitumor activity [17]. At ESMO 2022, extended follow-up data for a larger cohort of patients with selected tumor types were presented [18].
As of June 30, 2022, 147 pretreated patients received DS-7300 (IV, Q3W) at doses of 4.8 mg/kg to 16.0 mg/kg in the dose escalation part I of this study. In part II (dose expansion), 66 eligible patients with advanced/unresectable or metastatic solid tumors (unselected for B7-H3 expression) divided into three cohorts (cohort 1, patients with esophageal squamous cell cancer [ESCC]; cohort 2, patients with metastatic castration-resistant prostate cancer [mCRPC]; cohort 3, patients with squamous non-small cell lung cancer [sqNSCLC]) received the DS-7300 recommended dose of 12.0 mg/kg (IV, Q3W) as monotherapy. The key primary endpoint was DLTs, serious AEs (SAEs), TEAEs and AE of interest (AESI) in the dose escalation of the study, while ORR according to RECIST v1.1, duration of response (DoR), DCR, PFS and overall survival (OS) were analyzed in the dose expansion part.
In total, 35 patients (24 %) were still on treatment at the time those results were presented, including SCLC (40 %), mCRPC (23 %), ESCC (15 %), and sqNSCLC (56 %). Most patients showed a durable disease and tumor shrinkage across all tumor types and doses analyzed (Figure 3). Responses were observed in 32 % (95 % CI, 24-41).
For the SCLC subset, after a median follow-up of 4.9 months, a response was seen in 58 % (95 % CI, 33-80), the median time to response (TTR) was 1.2 months (95 % CI, NA-1.4), and the median DoR was 5.5 months (95 % CI, 2.8-NR). With respect to the mCRPC subgroup, after a median follow-up of 9.3 months, a response was measured in 33 % (95 % CI, 21-47) with 18 patients (33 %) achieving a PR; in this subgroup, the median DoR was 4.4 months (95 % CI, 2.7-NR), while seven responders remained on treatment. In the ESCC subset, after a median follow-up of 7.7 months, a response was detected in 23 % (95 % CI, 8-45) and half of the patients with post-baseline scans showed a tumor shrinkage; the median DoR was 2.8 months (95 % CI, 2.6-NR), with two responders still on treatment. After a median follow-up of 1.7 months for the sqNSCLC subset, a response was seen in 40 % (95 % CI, 5-85), with four out of five patients (80 %) having a post-baseline reduction in target lesions; the median DoR in this subgroup was 4.3 months (95 % CI, 3.1-NR) and one responder remained on treatment.
Regarding safety, the most common grade ≥ 3 TEAEs were anemia (19 %), neutropenia (4 %), nausea (3 %), pneumonia (3 %), and a decreased neutrophil count (3 %). A total of nine patients experienced interstitial lung disease (ILD) or pneumonitis, of which seven were allocated as drug-related ILD (grade 1, n = 2; grade 2, n = 4; grade 5, n = 1), and two ILD or pneumonitis events were pending adjudication.
The investigational B7-H3–directed DXd-ADC DS-7300 continues to show an encouraging antitumoral activity in heavily pretreated patients in several cancer types, including SCLC, mCRPC, ESCC, and sqNSCLC. No new safety signals were detected. These data further supported clinical development of DS-7300, including a phase II dose-optimization study in SCLC (NCT05280470), the design and current status of which was presented at ESMO 2022, too [19].
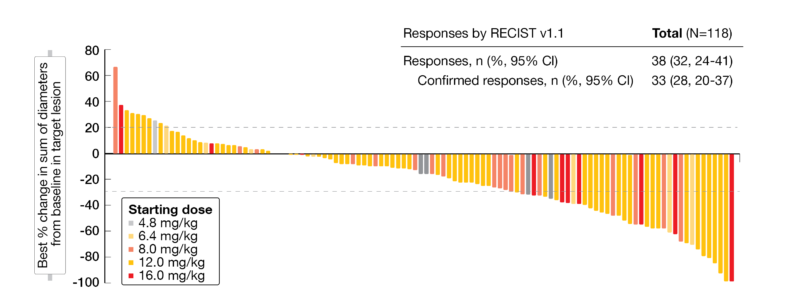
Figure 3: Waterfall-plot indicating the best percentage of change from baseline for target lesions.
Phase I trial of IMC-F106C in selected advanced solid tumors
Immune-mobilizing monoclonal T-cell receptors against cancer (ImmTAC) are a new class of T-cell–redirecting bispecific fusion proteins that use an engineered high-affinity T-cell receptor to target any protein, including intracellular antigens, that is presented as a peptide–HLA complex on the surface of target cells. ImmTAC molecules previously showed an OS benefit in uveal melanoma (HR, 0.51) [20]. The investigational agent IMC-F106C was designed for the treatment of tumors positive for the tumor-associated antigen PRAME. This first-in-human trial evaluates the safety and efficacy of IMC-F106C in adult patients who have the appropriate HLA-A2 tissue marker and whose cancer is positive for PRAME [21]. At ESMO 2022, results from the phase I dose escalation study (NCT04262466) of IMC-F106C in patients with selected advanced solid tumors were discussed [22].
Patients with advanced solid tumors positive for HLA-A*02:01 according to central testing and PRAME as confirmed by immunohistochemistry (IHC) received IMC-F106C weekly (IV) with intra-patient dose escalation (0.3 to 320 µg) over the first three weeks. Tumor assessment took place every nine weeks. The primary endpoint was to determine the expansion dose, while the preliminary antitumor activity, pharmacokinetic and pharmacodynamic markers were analyzed secondarily.
Pharmacologically, IMC-F106C showed a strong and consistent pharmacodynamic activity at 20 µg or higher; indeed, in peripheral blood, strong interferon gamma (IFNγ) induction and high T-cell trafficking (22-fold increase) were detected. Median PRAME H-score in the efficacy population was high (90 %). Responses to IMC-F106C were observed in multiple tumor types: three out of six (50 %) uveal melanoma patients, two out of six (33 %) cutaneous melanoma patients and two out of four (50 %) serous ovarian patients showed a partial response (one of them unconfirmed). Most patients had a durable tumor response or disease stabilization, with six out of the seven observed partial responses still ongoing at the time of data presentation at ESMO 2022 and two patients showing an ongoing PR for more than seven months. Patients with lower PRAME expression seemed to progress earlier, whereas high PRAME expression was associated with response and benefit. Moreover, out of 20 patients with available circulating tumor DNA (ctDNA), four achieved a PR with a tumor reduction greater of 50 %, including three with a clearance.
In relation to safety, IMC-F106F was well tolerated. The most reported AEs (all grades) were pyrexia (56 %) and cytokine release syndrome (49 %), the most frequent grade ≥3 AE being lymphopenia (15 %). No treatment-related discontinuation or grade 5 AE were reported.
IMC-F106C showed durable responses across multiple tumor types with a manageable safety profile. The dose escalation continues, while combinations with chemotherapy and checkpoint inhibitors are planned.
ACTIVATE trial: etigilimab combined to nivolumab
TIGIT is an inhibitory immunoreceptor that is upregulated by immune cells, like activated T‑cells and natural killer (NK) cells, as well as regulatory T‑cells in several cancer entities [23]. Etigilimab (MPH313) is a FCγR competent, humanized anti-TIGIT IgG1 monoclonal antibody that inhibits its interaction with PVR (poliovirus receptor) and therefore inhibits downstream signaling. In an early clinical trial (phase Ia/b), etigilimab showed an acceptable safety profile and antitumor activity either as monotherapy or in combination with nivolumab [24]. At this year’s ESMO meeting, an interim biomarker analysis of the ACTIVATE study (NCT04761198) combining etigilimab (etig) with the anti-PD1 nivolumab (nivo) was presented [25].
In this open-label phase Ib/II basket study, the efficacy, safety, tolerability, and pharmacokinetics/pharmacodynamics of etig plus nivo have been evaluated in selected locally advanced or metastatic solid tumors. This study included a biomarker monitoring to follow the activation of immunological modulators by flow cytometry from peripheral blood; additionally, plasma samples were also analyzed for cytokine changes and ctDNA. At baseline, tissue samples (FFPE) were collected to determine PD-L1 expression by cIHC or multiplex immune-fluorescence (F-IHC) and mRNA seq.
No objective responses were observed in PVR low and TIGIT negative tumors patients. On the contrary, patients showing a TIGIT-high tumor expression had a clinical benefit (58 % in TIGIT-high patients vs 33 % in TIGIT-low patients) and ORR (33 % vs 10 %, respectively). Moreover, in patients with a clinical benefit, a robust target engagement was observed though increases in proliferating CD4 and CD8 effector memory populations, as well as NK cells and PD-1-positive T-cells. Furthermore, an increase in IFNγ was observed in NK cells and CD4 EM T-cells as well as an elevation of the IL-2 production in CD4 EM T cells. Tissue biomarker (PVR, CD226/CD8) expression at baseline was associated with tumor shrinkage (2 patients with a CR and 5 patients with a PR). A decrease of ctDNA at five to six weeks post-treatment correlated with an objective response.
These interim biomarker data from Etig plus nivo combination therapy brought evidence of a dual TIGIT/PD-1 blockade and was associated with increased levels of proliferating and cytokine producing T-cells in circulation. Furthermore, ctDNA reductions correlated with clinical response. Altogether, these preclinical data support further evaluation of PVR, TIGIT and CD226 as potential biomarkers for this therapeutic combination.
REFERENCES
- Reinhard, K, et al., An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020; 367(6476): 446-453.
- Mackensen, A, et al., BNT211-01: A Phase 1 trial to evaluate safety and efficacy of CLDN6 CAR T cells and CLDN6-encoding mRNA vaccine-mediated in vivo expansion in patients with CLDN6-positive advanced solid tumours. Ann Oncol 2022; 33(suppl 7; Abstr LBA38).
- EMA, Recommendations on eligibility to PRIME scheme. 2022 last accessed on October 16, 2022; Available from: https://www.ema.europa.eu/en/documents/chmp-annex/recommendations-eligibility-prime-scheme-adopted-chmp-meeting-20-23-june-2022_en-0.pdf.
- Carvalho, T, mRNA vaccines boost BioNTech’s CAR T cell therapy. Nat Med 2022. Epub ahead a print.
- Wischhusen, J, et al., Growth/Differentiation Factor-15 (GDF-15): From Biomarker to Novel Targetable Immune Checkpoint. Front Immunol 2020; 11: 951.
- Looi, CK, et al., The Role of Ras-Associated Protein 1 (Rap1) in Cancer: Bad Actor or Good Player? Biomedicines 2020; 8(9): 334.
- Melero, I, et al., Final results of the first-in-human clinical trial of the GDF-15 neutralizing antibody CTL-002 in combination with nivolumab in subjects with solid tumors relapsed/refractory to prior anti-PD1/PD-L1 treatment. Ann Oncol 2022; 33(suppl 7; Abstr 729MO).
- Zhao, Y, et al., The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim Biophys Sin (Shanghai) 2014; 46(3): 180-189.
- Shi, D, et al., Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer 2012; 3(3-4): 240-248.
- Momand, J, et al., The MDM2 gene amplification database. Nucleic Acids Res 1998; 26(15): 3453-3459.
- Cornillie, J, et al., Anti-tumor activity of the MDM2-TP53 inhibitor BI-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring MDM2 amplification. Clin Transl Oncol 2020; 22(4): 546-554.
- Schoeffski, P, et al., A phase I dose-escalation and expansion study evaluating the safety and efficacy of the MDM2–p53 antagonist BI 907828 in patients (pts) with solid tumours. Ann Oncol 2022; 33(suppl 7; Abstr 452O).
- Picarda, E, et al., Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin Cancer Res 2016; 22(14): 3425-3431.
- Dong, P, et al., B7H3 As a Promoter of Metastasis and Promising Therapeutic Target. Front Oncol 2018; 8: 264.
- Kontos, F, et al., B7-H3: An Attractive Target for Antibody-based Immunotherapy. Clin Cancer Res 2021; 27(5): 1227-1235.
- Nakada, T, et al., The Latest Research and Development into the Antibody-Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chem Pharm Bull (Tokyo) 2019; 67(3): 173-185.
- Johnson ML., A phase I/II multicenter, first-in-human study of DS-7300 (B7-H3 DXd-ADC) in patients (pts) with advanced solid tumors. Ann Oncol 2021; 32(suppl 5; Abstr 513O).
- Doi, T, et al., DS-7300 (B7-H3 DXd antibody-drug conjugate [ADC]) shows durable antitumor activity in advanced solid tumors: Extended follow-up of a phase I/II study. Ann Oncol 2022; 33(suppl 7; Abstr 453O).
- Paz-Ares, L, et al., Phase II, multicenter, randomized, open-label study of DS-7300 in patients (pts) with pre-treated extensive-stage small cell lung cancer (ES-SCLC). Ann Oncol 2022; 33(suppl 7; Abstr 1550TiP).
- Nathan, P, et al., Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N Engl J Med 2021; 385(13): 1196-1206.
- Moureau, S, et al., IMC-F106C, a novel and potent immunotherapy approach to treat PRAME expressing solid and hematologic tumors. Cancer Res 2020; 80(suppl 16; Abstr 5572).
- Hamid O., et al., Results from phase I dose escalation of IMC-F106C, the first PRAME × CD3 ImmTAC bispecific protein in solid tumors. Ann Oncol 2022; 33(suppl 7; Abstr 728O).
- Chauvin, JM, et al., TIGIT in cancer immunotherapy. J Immunother Cancer 2020; 8(2): e000957.
- Mettu, NB, et al., A Phase 1a/b Open-Label, Dose-Escalation Study of Etigilimab Alone or in Combination with Nivolumab in Patients with Locally Advanced or Metastatic Solid Tumors. Clin Cancer Res 2022; 28(5): 882-892.
- Sarikonda, G, et al., Interim biomarker analysis of a phase Ib/II study of anti-TIGIT etigilimab (MPH313) and nivolumab in subjects with select locally advanced or metastatic solid tumors (ACTIVATE). Ann Oncol 2022; 33(suppl 7; Abstr 111P).
© 2022 Springer-Verlag GmbH, Impressum
More posts
Promising therapeutic strategies for colorectal cancer treatment
Promising therapeutic strategies for colorectal cancer treatment With more than
From neoadjuvant to second-line therapeutic options for RCC patients
From neoadjuvant to second-line therapeutic options for RCC patients Kidney can
Towards treating all stages of gastric cancer and gastroesophageal junction adenocarcinoma
Towards treating all stages of gastric cancer and gastroesophageal junction adenoca
Update on the treatment for advanced HCC
Update on the treatment for advanced HCC With more than 900,000 newly diagnosed
New clinical insights in head and neck squamous cell carcinoma
New clinical insights in head and neck squamous cell carcinoma Head and neck squamous c
Preface – ESMO Solid Tumor 2022
Preface – ESMO Solid Tumors 2022 © privat - Jaime R. Merchan, MD. MMSc, University of