Promising therapeutic strategies for colorectal cancer treatment
With more than 1.9 million new cases, colorectal cancer (CRC) accounted in 2020 for 10 % of all diagnosed cancers. CRC ranked third in terms of incidence and was the second leading cause of cancer mortality worldwide, with 935,000 estimated deaths in 2020 [1]. Over the last five years, clinical studies have shown that treatments specifically tailored to the molecular and pathological characteristics of the tumor improved overall survival. In addition, genomic profiling able to detect somatic variants represents an important asset to identify effective treatments for specific patient subsets [2]. At this year’s ESMO meeting, the recent updates on CRC treatment were presented.
Neoadjuvant immune-checkpoint inhibition in dMMR CRC
Deficient mismatch repair (dMMR) occurs in approximately 10-15 % of colon cancers, with one-third of dMMR CRC being associated with the Lynch Syndrome [3]. Despite long established standard-of-care chemotherapy, recurrence rates of stage III dMMR tumors are still 20-40 % and closely associated with a poor survival [4]. Recent studies have shown that neoadjuvant immunotherapy leads to a clinically meaningful pathological response in various malignancies – such as bladder cancer or melanoma – and was associated with excellent outcomes [5, 6]. NICHE-1 was the first study with neoadjuvant immunotherapy in non-metastatic dMMR colon cancer where all patients had a pathological response and 60 % a pathologic complete response [7].
At ESMO 2022, Chalabi et al. presented the first results of the NICHE-2 trial (NL58483.031.16, EudraCT 2016-002940-17), a non-randomized study in patients with non-metastatic, untreated dMMR colon adenocarcinoma [8]. Overall, 112 eligible patients were treated with one dose of ipilimumab (1 mg/kg) plus nivolumab (3 mg/kg) in the first cycle, then only nivolumab in the second cycle two weeks later, followed by surgery within six weeks of enrollment. The co-primary endpoints were safety, feasibility, and 3-year disease-free survival (DFS).
Among 112 patients enrolled in this study, 107 were available for the efficacy analysis; 74 % had high-risk stage III CRC, including 35 % and 28 % patients with clinical T4a and T4b tumors, respectively. In total, 98 % of patients underwent timely surgery, with a median time between first dose and surgery of 5.4 weeks, thus meeting the primary safety endpoint. Overall, 99 % of patients achieved a pathologic response, with a pathologic complete response (pCR, defined as 0 % residual viable tumor) observed in 67 % of patients, a major pathologic response (MPR, defined as ≤ 10 % residual viable tumor) in 95 % of them and a partial pathologic response (pPR, defined as 10 - 50 % of residual viable tumor) in 4 % (Figure 1). The rate of pCR was higher in patients with Lynch syndrome (n = 32) than in those with sporadic tumors (n = 65) (78 % vs 58 %; p = 0.056). After a median follow-up of 13.1 months, no disease recurrence has been observed in any patient, which might predict promising upcoming results. Moreover, the treatment was well tolerated, with only 4 % grade ≥ 3 immune-related adverse events. Further results on 3-year DFS are expected in 2023.
These initial results from the NICHE-2 trial demonstrated that neoadjuvant immunotherapy consisting of ipilimumab plus nivolumab has the potential to become a new standard-of-care in dMMR colon cancer.

Figure 1: Pathologic response in the NICHE-2 trial. PR, pathologic response; MPR, major pathologic response
Update on KRYSTAL-1: adagrasib ± cetuximab in KRASG12C-mutated CRC
KRASG12C mutations, which are reported in approximately 3 % of CRC tumors, are associated with a poor prognosis [9]. They are a negative predictor of cetuximab – an anti-EGFR monoclonal antibody – efficacy, and late treatment options are limited [10]. Adagrasib, an irreversible and selective KRASG12C inhibitor, has been optimized with favorable pharmacokinetic properties, including a long half-life (23 h), extensive tissue distribution, dose-dependent pharmacokinetics, and central nervous system (CNS) penetration [11, 12]. Thus, the combination of cetuximab with adagrasib may enhance the inhibition of KRAS-dependent signaling or overcome adaptive feedback and provide a more durable efficacy.
KRYSTAL-1 (NCT03785249) is a phase Ib/II multicohort study evaluating adagrasib as monotherapy or in combinations with cetuximab across patients with advanced solid tumors harboring a KRASG12C mutation [13]. In the CRC cohort, eligible patients received either adagrasib (600 mg, orally, twice daily [BID]) plus cetuximab (400 mg/m2, followed by 250 mg/m2 once weekly [QW]or 500 mg/m2 every other week [Q2W]) in phase Ib or adagrasib monotherapy in phase II. Primary study endpoints included safety, recommended phase II dose and pharmacokinetics in phase Ib and overall response rate (ORR) according to RECIST v1.1 in phase II.
At ESMO 2022, Klempner et al. reported updated results from CRC cohorts (phase Ib and II), with a median follow-up of 17.5 and 20.1 months, respectively [14]. As of June 16, 2022, 44 patients received adagrasib monotherapy and 32 patients received adagrasib plus cetuximab. Baseline characteristics were balanced between both cohorts, with a median of three prior lines of systemic therapy each. Forty-three patients with adagrasib monotherapy and 28 patients on combination therapy were evaluable for efficacy. At the time of data cut-off, a clinical benefit was observed for the combination therapy, in terms of confirmed ORR (46 % for adagrasib plus cetuximab vs 19 % for adagrasib) (Figure 2), disease control rate (DCR, 100 % vs 86 %), median duration of response (DoR, 7.6 vs 4.3 months), progression-free survival (PFS, 6.9 vs 5.6 months) and median overall survival (OS, 13.4 vs 19.8 months), with a 1-year survival rate of 69 % versus 61 %, respectively. Tumor shrinkage of any extent was observed in 93 % of patients treated with the combined therapy versus 79 % of those who were administered adagrasib monotherapy.
Adagrasib had a manageable toxicity, as grade ≥3 TRAEs were experienced by 30 % of patients in the monotherapy arm (mostly anemia [9 %] and diarrhea [7 %]) and by 9 % of patients in the combination arm (diarrhea, dermatitis acneiform and stomatitis, 3 % each). No serious TRAEs were observed.
So far, the KRYSTAL-1 trial showed an encouraging clinical activity of adagrasib both as monotherapy and in combination with cetuximab particularly in heavily pretreated patients with KRASG12C-mutated CRC. This therapeutic combination is being currently evaluated in the phase III KRYSTAL-10 trial (NCT04793958) in the second line (2L) setting of this patient population.
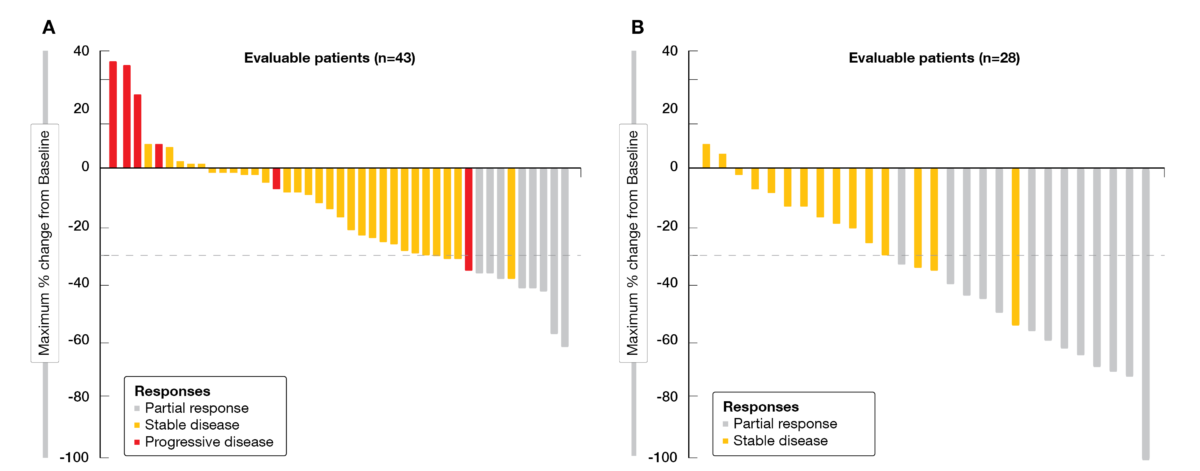
Figure 2: Waterfall plots of best tumor change from baseline with adagrasib monotherapy (A) or adagrasib plus cetuximab (B) in the KRYSTAL-1 trial.
CodeBreaK 101: sotorasib plus panitumumab
In the phase II CodeBreaK 100 trial (NCT03600883), sotorasib – a KRASG12C inhibitor – monotherapy was previously shown to elicit modest benefit in heavily pretreated patients with KRASG12C-mutated CRC [15]. Unfortunately, resistance to sotorasib may occur as a feedback reactivation of the RAS-MAPK pathway and accumulation of activated EGFR [16, 17]. Initial data from the phase I CodeBreaK 101 trial (NCT04185883) have shown that sotorasib combined with the EGFR antibody panitumumab appears to inhibit tumor growth to a better extend than sotorasib monotherapy in patients with chemorefractory KRASG12C-mutated metastatic CRC (mCRC) [18, 19]. Safety and efficacy data of the combination therapy from the fully enrolled dose expansion cohort (n = 40) were presented by Kuboki et al. at ESMO 2022 [20].
Inclusion criteria in the CodeBreaK 101 study comprised KRASG12C inhibitor-naïve patients who were diagnosed with KRASG12C-mutated mCRC through molecular testing, at least one prior treatment for advanced disease, and a progression on or after fluoropyrimidine, oxaliplatin, irinotecan, or an anti-angiogenic agent. Patients in the dose-expansion cohort received 960 mg of sotorasib (960 mg, orally, daily) and panitumumab (6 mg/kg, intravenous [IV], every second week [Q2W]). Primary endpoints enclosed safety and tolerability, while secondary endpoints included ORR, DCR, DoR, time to response (TTR), PFS per RECIST v1.1, OS, and pharmacokinetics.
Overall, 40 patients with a median age of 58 years were enrolled in this study. Most patients were heavily pretreated, with a median of two prior lines of therapy. A confirmed ORR of 30 % (95 % CI, 16.6-46.5), was obtained, with all responders (n = 12) showing a partial response. A stable disease (SD) was determined in 25 patients (63 %), the DCR reached 93 % (95 % CI, 79.6-98.4). Moreover, tumor shrinkage of the target lesions according to RECIST v1.1 was observed in 87.5 % of patients. The median duration of treatment was 5.9 months, with 25 % of patients remaining on treatment at data cut-off (June 24, 2022). After a median follow-up of eleven months, the median DOR reached 4.4 months (range, 2.8-7.4) and the median PFS was 5.7 months (95 % CI, 4.2-7.6). Of note, the 9-month PFS rate attained 12.3 % (95 % CI, 3.4-27.2). The median OS was not reached at the time of analysis, but the 9-month OS rate attained 82.5 % (95 % CI, 61.8-92.6).
The combination was very well tolerated, with TRAEs of grade 3 occurring in 23 % of patients and no grade 4 or fatal TRAEs. No patient had to discontinue either drug due to TRAEs. Dose interruptions or reductions due to TRAEs, were related to sotorasib treatment in 15 % of patients and associated with panitumumab therapy in 25 %.
This updated analysis of the CodeBreak 101 dose expansion cohort provides further evidence that sotorasib plus panitumumab is safe and tolerable in chemorefractory patients with KRASG12C-mutated mCRC. Compared to previously reported sotorasib monotherapy outcomes, the threefold higher response obtained with the sotorasib plus panitumumab supports further development of this new therapeutic approach. Thus, the currently ongoing global phase III CodeBreaK 300 trial (NCT05198934) is evaluating sotorasib plus panitumumab versus standard therapy of the investigator’s choice for the treatment of KRASG12C-mutated mCRC.
FRESCO-2: fruquintinib in refractory mCRC
The vascular endothelial growth factor (VEGF) signaling pathway is a key mediator of angiogenesis, which is involved in tumor growth and metastasis [21]. Fruquintinib is a highly selective and potent oral tyrosine kinase inhibitor (TKI) of VEGF receptor (VEGFR) 1, 2, and 3 [22]. In the phase III FRESCO trial (NCT02314819), fruquintinib already demonstrated efficacy and safety in Chinese patients with mCRC in the third-line setting and beyond (3L+), which led to its approval in China in 2018 [23]. To better reflect current global treatment practice, the phase III FRESCO-2 study (NCT04322539) evaluated the antitumoral activity and tolerability of fruquintinib in heavily pretreated mCRC patients from the United States, Europe, Japan, and Australia. The results of the FRESCO-2 trial were presented by Dasari et al. at ESMO 2022 [24].
The study design of the FRESCO-2 trial required eligible patients to have previously received a fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, an anti-VEGF therapy, and an anti-EGFR therapy for RAS wild-type, as well as a prior treatment with an immune checkpoint inhibitor (ICI) or BRAF inhibitor if indicated. They also had to have progression on or intolerance to TAS-102 and/or regorafenib. Patients were randomized 2:1 to receive either fruquintinib (5 mg orally, once daily [QD] for three weeks, followed by one week off) or placebo until progression or unacceptable toxicity. All patients were additionally administered best supportive care. The primary endpoint was OS, while PFS, ORR, DCR, and safety were secondarily analyzed.
Overall, 691 patients (fruquintinib, n = 461; placebo, n = 230) were randomized between September 2020 and December 2021 in this study. Patients were heavily pretreated, with a median of five prior lines of treatment in both arms. After a median follow-up of 11.3 months for fruquintinib and 11.2 months for placebo (data cut-off of June 24, 2022), the study met its primary endpoint in terms of median OS improvement (7.4 months in the fruquintinib arm vs 4.8 months with placebo; HR, 0.662; 95 % CI, 0.549-0.800; p < 0.001), and therefore a 33.8 % risk reduction of death (Figure 3). The median PFS was also in favor of fruquintinib (3.7 months vs 1.8 months, respectively; HR, 0.321; 95 % CI, 0.267-0.386; p < 0.001); this PFS-improvement was consistent across the pre-specified subgroups. The DCR was significantly higher in the fruquintinib arm compared to placebo (55.5 % vs 16.1 % with an adjusted difference of 39.4 % (95 % CI, 32.8-46.0; 2-sided p < 0.001) and the ORR reached 1.5 % versus 0 % (2-sided p = 0.059), respectively.
In terms of safety profile, more grade ≥ 3 treatment-related adverse events (TEAEs) were reported by patients who received fruquintinib compared to placebo (62.7 % vs. 50.4 %), most frequently hypertension (13.6 % vs 0.9 %), asthenia (7.7 % vs 3.5 %) and hand-foot syndrome (6.4 % vs 5.3 %). In both arms, the proportion of any serious TEAEs was similar (37.5 % vs 38.3 %).
The FRESCO-2 trial met its primary endpoint, OS, and its key secondary endpoint, PFS, overall showing that fruquintinib therapy resulted in a significant and clinically meaningful improvement in patients with refractory mCRC. The results are consistent with those of the prior FRESCO trial and support fruquintinib as a new treatment option in this patient population.

Figure 3: Overall survival according to treatment received in der FRESCO-2 study (ITT population).
ERMES trial: maintenance therapy with cetuximab ± FOLFIRI
Cetuximab, a highly selective antibody against EGFR, has previously demonstrated its efficacy and safety in combination with FOLFIRI (leucovorin, fluorouracil [5-FU] and irinotecan) as first-line treatment in EGFR-expressing, RAS-wt mCRC [25]. Although maintenance therapy following anti-EGFR based treatment is controversial and evidence lies on three phase II studies (MACRO 2, VALENTINO, PANAMA), the phase III EREMES trial (NCT02484833) is currently investigating whether a maintenance with cetuximab monotherapy after FOLFIRI plus cetuximab induction is a valid choice in RAS/BRAF-wt mCRC. Initial results were presented by Armando Orlandi at this year’s ESMO meeting [26].
In the ERMES study, it was investigated whether cetuximab alone (arm B, given until progression or cumulative toxicity) after eight cycles of FOLFIRI plus cetuximab results in a non-inferiority when compared with continuous FOLFIRI plus cetuximab (arm A, given until progression or cumulative toxicity) in untreated RAS/BRAF-wt mCRC patients who were randomized 1:1. Co-primary endpoints of this study were PFS for the modified per-protocol (mPP) population (treated beyond cycle 8) per blinded independent central review (BICR) and safety in term of incidence of G3-4 AE. Secondary endpoints included PFS in the modified intention-to-treat (mITT) population (who received at least one cycle of treatment), OS in the mPP and mITT populations, ORR, and quality of life [27].
A total of 606 patients were randomized between May 2015 and March 2020; a drop-out rate of 40 % left the mPP population at only 337 patients (arm B, n = 183; arm A, n =154;). With a median follow-up of 22.3 months, the median PFS of the mPP population failed to demonstrate a non-inferiority for maintenance therapy with cetuximab monotherapy (arm B, 10.0 months vs arm A, 12.2 months; HR, 1.30; 95 % CI, 1.03-1.64; p = 0.43). To note, in the mPP population, the median PFS of patients with right-sided primary tumor location showed a significant benefit of continuous FOLFIRI plus cetuximab (arm B, 8.29 months vs arm A, 11.73 months; HR, 2.07; 95 % CI, 1.20-3.57; p = 0.007). In the mITT population, median PFS was 9.01 months versus 10.72 months, respectively (HR, 1.1; 95 % CI, 0.92-1.31; p = 0.305). The median OS was 36.64 months in arm B versus 30.76 months in arm A in the mPP population (HR, 0.81; 95 % CI, 0.80-1.09; p = 0.157), and 31.09 months vs 25.36 months in the mITT population (HR, 0.9; 95 % CI, 0.72-1.12; p = 0.327). In the mPP population, the ORR was higher in arm B (71.6 %; 95 % CI, 64.5-78.0, including 8.8 % of patients with a CR) compared to arm A (67.5 %; 95 % CI, 59.5-74.9, including 10.5 % of patients with a CR).
Grade 3-4 adverse events – the co-primary endpoint – occurred in 39.9 % of patients in arm B and 44.2 % of patients in arm A in the whole treatment period. The most frequent grade 3-4 AEs were skin disorders (18.0 % in arm B vs 20.1 % in arm A), a decreased neutrophil count (9.8 % vs 14.9 %) and diarrhea (8.2 % vs 11.0 %).
The EREMES trial failed to demonstrate non-inferiority of the maintenance therapy with cetuximab monotherapy, but the results may have been negatively influenced by the unexpectedly high dropout rate. Nevertheless, the OS analysis of the mPP and mITT populations showed promising results still supporting the hypothesis of a de-escalation therapy with cetuximab monotherapy as an option for selected RAS/BRAF-wt CRC patients. In this way, the pre-planned translational research on tissue sample and liquid biopsies with NGS is ongoing.
Resistance alterations in BRAF V600E-mutated mCRC
The BRAF V600E mutation is associated with a poor prognosis for patients with mCRC and the inhibition of BRAF alone has limited efficacy due to the pathway reactivation through EGFR signaling. In this context, the BEACON trial (NCT02928224), evaluating the triple combination of encorafenib (a BRAF inhibitor) plus binimetinib (a MEK inhibitor) plus cetuximab (a EGRF inhibitor), demonstrated an improved OS compared to doublet therapy (encorafenib + cetuximab) or standard chemotherapy (FOLFIRI + cetuximab or irinotecan + cetuximab) in patients with previously treated BRAF V600E-mutated mCRC [28]. To identify genomic alterations possibly mediating treatment resistance, plasma samples were collected from study participants at baseline and at the end of the treatment (EOT). Circulating tumor DNA (ctDNA) was analyzed by genomic profiling using GuardantOMNITM. Somatic alterations, including non-synonymous mutations, amplifications, and rearrangement, as well as other patient-level genomic alterations, were evaluated to characterize acquired alterations [29]. At ESMO 2022, Kopetz et al. presented the results of this genomic post-hoc analysis [30].
To investigate mechanisms of resistance, paired baseline and EOT samples were evaluated from 320 out of the 665 patients (48.1 %) enrolled in the BEACON trial. The most frequently acquired resistant alterations in both, the triplet therapy (encorafenib + binimetinib + cetuximab) and the doublet therapy (encorafenib + cetuximab), were mutations in KRAS (40.2 % and 44.6 %), NRAS (25.0 % and 36.6 %) and amplification of MET (19.6 % and 17.0 %) compared with the control arm (FOLFIRI plus cetuximab or irinotecan plus cetuximab, [0 %, 3.2 % and 0 %], respectively). MAP2K1 (MEK1) mutations were observed in more doublet-treated patients than triplet-treated patients (16.1 % vs 3.6 %; p < 0.01). At least one of the above-mentioned mutations, and/or MET, KRAS, BRAF, and IGFR1 per patient, was found in patients treated with triplet-, doublet-, or control-treated patients (62.5 %, 63.4 , 7.4 %, respectively).
This post-hoc analysis of the BEACON trial showed that in patients with previously treated BRAF V600E-mutated mCRC, MAPK pathway reactivation is a common mechanism of resistance following inhibition of BRAF and EGFR ± MEK inhibition. Moreover, enhanced receptor signaling was more prevalent in the triplet arm, whereas acquired mutations in RAS-MEK signaling were more prevalent in the doublet arm.
REFERENCES
- Sung, H, et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021; 71(3): 209-249.
- Biller, LH, et al., Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 2021; 325(7): 669-685.
- De’ Angelis, GL, et al., Microsatellite instability in colorectal cancer. Acta Biomed 2018; 89(9-s): 97-101.
- Andre, T, et al., Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J Clin Oncol 2015; 33(35): 4176-4187.
- Einerhand, SMH, et al., Survival after neoadjuvant/induction combination immunotherapy vs combination platinum-based chemotherapy for locally advanced (Stage III) urothelial cancer. Int J Cancer, epub ahead a print 2022.
- Menzies, AM, et al., Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat Med 2021; 27(2): 301-309.
- Chalabi, M, et al., Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 2020; 26(4): 566-576.
- Chalabi, M, et al., Neoadjuvant immune checkpoint inhibition in locally advanced MMR-deficient colon cancer: The NICHE-2 study. Ann Oncol 2022; 33(suppl 7; Abstr LBA7).
- Henry, JT, et al., Comprehensive Clinical and Molecular Characterization of KRAS (G12C)-Mutant Colorectal Cancer. JCO Precis Oncol 2021; 5.
- Modest, DP, et al., Impact of the specific mutation in KRAS codon 12 mutated tumors on treatment efficacy in patients with metastatic colorectal cancer receiving cetuximab-based first-line therapy: a pooled analysis of three trials. Oncology 2012; 83(5): 241-247.
- Hallin, J, et al., The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020; 10(1): 54-71.
- Janne, PA, et al., Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRAS(G12C) Mutation. N Engl J Med 2022; 387(2): 120-131.
- Weiss, J, et al., KRYSTAL-1: Adagrasib (MRTX849) as monotherapy or combined with cetuximab (Cetux) in patients (Pts) with colorectal cancer (CRC) harboring a KRASG12C mutation. Ann Oncol 2022; 33(suppl 5; Abstr LBA6).
- Klempner, SJ, et al., KRYSTAL-1: Updated efficacy and safety of adagrasib (MRTX849) with or without cetuximab in patients with advanced colorectal cancer (CRC) harboring a KRASG12C mutation. Ann Oncol 2022; 33(suppl 7; Abstr LBA24).
- Fakih, MG, et al., Sotorasib for previously treated colorectal cancers with KRAS(G12C) mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol 2022; 23(1): 115-124.
- Amodio, V, et al., EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov 2020; 10(8): 1129-1139.
- Ryan, MB, et al., KRAS(G12C)-independent feedback activation of wild-type RAS constrains KRAS(G12C) inhibitor efficacy. Cell Rep 2022; 39(12): 110993.
- Canon, J, et al., The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019; 575(7781): 217-223.
- Lou, K, et al., KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal 2019; 12(583).
- Kuboki, Y, et al., Sotorasib in combination with panitumumab in refractory KRAS G12C-mutated colorectal cancer: Safety and efficacy for phase Ib full expansion cohort. Ann Oncol 2022; 33(suppl 7; Abstr 315O).
- Hicklin, DJ, et al., Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 2005; 23(5): 1011-1027.
- Sun, Q, et al., Discovery of fruquintinib, a potent and highly selective small molecule inhibitor of VEGFR 1, 2, 3 tyrosine kinases for cancer therapy. Cancer Biol Ther 2014; 15(12): 1635-1645.
- Li, J, et al., Effect of Fruquintinib vs Placebo on Overall Survival in Patients With Previously Treated Metastatic Colorectal Cancer: The FRESCO Randomized Clinical Trial. JAMA 2018; 319(24): 2486-2496.
- Dasari, NA, et al., FRESCO-2: A global phase III multiregional clinical trial (MRCT) evaluating the efficacy and safety of fruquintinib in patients with refractory metastatic colorectal cancer. Ann Oncol 2022; 33(suppl 7; Abstr LBA25).
- Heinemann, V, et al., FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol 2014; 15(10): 1065-1075.
- Pinto, C, et al., Phase III study with FOLFIRI/cetuximab versus FOLFIRI/cetuximab followed by cetuximab (Cet) alone in first-line therapy of RAS and BRAF wild-type (wt) metastatic colorectal cancer (mCRC) patients: The ERMES study. Ann Oncol 2022; 33(suppl 7; Abstr LBA22).
- Falter, T, et al., Depression and cognitive deficits as long-term consequences of thrombotic thrombocytopenic purpura. Transfusion 2017; 57(5): 1152-1162.
- Tabernero, J, et al., Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J Clin Oncol 2021; 39(4): 273-284.
- Kopetz, S, et al., Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N Engl J Med 2019; 381(17): 1632-1643.
- Kopetz, S, et al., Genomic mechanisms of acquired resistance of patients (pts) with BRAF V600E-mutant (mt) metastatic colorectal cancer (mCRC) treated in the BEACON study. Ann Oncol 2022; 33(suppl 7; Abstr 316O).
© 2022 Springer-Verlag GmbH, Impressum
More posts
Promising therapeutic strategies for colorectal cancer treatment
With more than 1.9 million new cases, colorectal cancer (CRC) accounted in 2020 for 10 % of all diagnosed cancers. CRC ranked third in terms of incidence and was the second leading cause of cancer mortality worldwide, with 935,000 estimated deaths in 2020. Over the last five years, clinical studies have shown that treatments specifically tailored to the molecular and pathological characteristics of the tumor improved overall survival.
From neoadjuvant to second-line therapeutic options for RCC patients
Kidney cancer accounted for more than 400,000 newly diagnosed cases in 2020,. Interestingly, after over two decades of increasing rates, the worldwide incidence of renal cell carcinoma (RCC) has shown signs of plateauing in recent years, whereas an increase of the global kidney cancer death rate was observed.
Towards treating all stages of gastric cancer and gastroesophageal junction adenocarcinoma
Gastric and esophagus cancers were the fifth and the seventh most frequently diagnosed cancers worldwide in 2020, respectively. In the same year, these cancers ranked fourth and sixth in mortality, respectively. Most patients with gastric cancer/gastroesophageal junction adenocarcinoma (GC/GEJA) present with inoperable or metastatic disease at the time of diagnosis; resulting in a strong need for efficient and tolerable first-line (1L) and second-line (2L) treatments.
Update on the treatment for advanced HCC
With more than 900,000 newly diagnosed cases in 2020, hepatocellular carcinoma (HCC) is the sixth most frequent malignant disease with a high mortality rate of approximately 92 %. Current first-line treatment for advanced HCC includes atezolizumab plus bevacizumab, as well as the tyrosine kinase inhibitors (TKIs) sorafenib and lenvatinib.
New clinical insights in head and neck squamous cell carcinoma
Head and neck squamous cell carcinoma (HNSCC) was the sixth most common cancer in 2018, with more than 700,000 newly diagnosed cases per year and 350,000 cancer deaths worldwide. Around 90 % of head and neck cancers are HNSCC, with oral cavity, oropharynx, hypopharynx, and larynx being the most commonly affected areas.
Preface – ESMO Solid Tumor 2022
At the ESMO congress held in Paris, France, and virtually from 9th – 13th September 2022, practice-changing data and high-quality education attracted more than 29,300 participants from over 150 countries.