

Overcoming resistance to targeted inhibitors
Subclonal mutations on covalent BTK inhibitor treatment
As covalent BTK inhibitors have been in use for the treatment of CLL in clinical practice for an extended period of time, different resistance mutations are being observed. Dr. Adrian Wiestner, MD, PhD, National Institutes of Health, Bethesda, USA, noted that mutations at progression are variable depending on the specific BTK inhibitor used, with the “classical” C481 mutations prevailing on ibrutinib and acalatinib treatment, while L528W mutations are mainly found in the context of zanubrutinib therapy. This variability is due to different chemical structures of BTK inhibitors that shape the BTK binding properties [1, 2].
CLL with primary resistance to ibrutinib treatment is virtually always transformed lymphoma (i.e., Richter’s transformation, Hodgkin lymphoma; Figure 1) [3]. Progression that occurs later on typically carries BTK or PLCG2 mutations. Dr. Wiestner presented unpublished findings according to which mutations are found in separate subclones and show different clonal evolution, with some undergoing linear evolution while others branch out in various subclones. The distribution of BTK and PLCG2 resistance mutations was demonstrated to differ across compartments such as peripheral blood, lymph nodes and bone marrow. Critical signaling pathways that had been downregulated at the time of response were restored in patients progressing on ibrutinib, with pathway activation being normalized to pre-treatment levels.
TP53 aberrations are common in patients progressing on BTK inhibitors. However, they do not represent a resistance mechanism, and successful long-term disease control with first-line ibrutinib is possible in the setting of TP53-aberrant CLL [4]. Similarly, deletion 8p is common in patients progressing on BTK inhibitor therapy and can already be present at baseline, but it only appears to set the stage for increased risk of the eventual acquisition of resistance mutations [5, 6]. The same applies to the immunologic marker CD49d, which is a prognostic factor for the time from diagnosis to treatment and a predictor for the efficacy of BTK inhibition [7, 8].
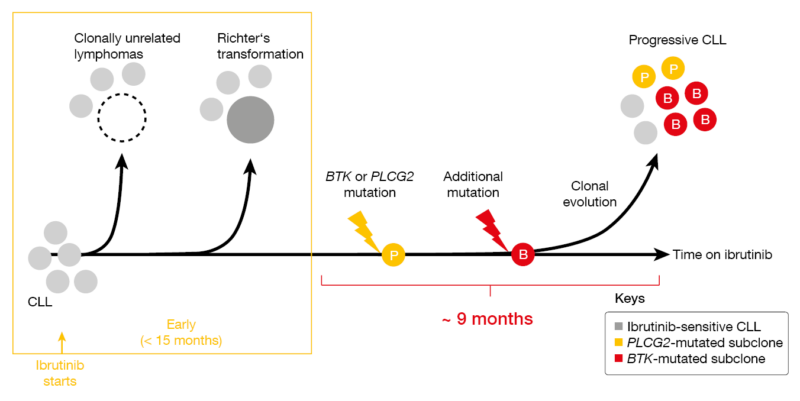
Figure 1: Clonal evolution observed in ibrutinib-treated patients with CLL
Non-covalent inhibition: pirtobrutinib
Data on the genomic evolution and resistance to the non-covalent BTK inhibitor pirtobrutinib were presented by Jennifer R. Brown, MD, PhD, Dana-Farber Cancer Institute and Harvard Medical School, Boston, USA. The BRUIN trial evaluated single-agent pirtobrutinib in 311 CLL patients; among these, 279 had previously received covalent BTK inhibitor treatment, and 111 showed progression on pirtobrutinib at data cutoff. For 49 of these, paired pre- and post-progression samples were available for targeted next generation sequencing (NGS).
At baseline, 51 % of patients had BTK mutations the vast majority of which were C481S mutations. At progression, 71 % had ≥ 1 acquired mutation. Acquired resistance to pirtobrutinib mainly converged around non-C481 BTK mutations, with the gatekeeper T474 mutation being most frequent. This was followed by the L528 mutation and other kinase mutations. Approximately half of patients did not acquire BTK mutations, and 29 % did not acquire any mutations, which suggests other mechanisms of resistance.
Decrease or clearance of C481 clones at the time of progression on pirtobrutinib were observed in 92 %, while BTK T474, L528, C481R/S/Y and other kinase mutations arose at or near progression. Importantly, baseline and acquired BTK mutations did not preclude pirtobrutinib efficacy. Overall response rates (ORRs) were high regardless of the type of acquired BTK mutation, ranging from 83 % to 92 %. Twenty-four percent of acquired non-C481 BTK mutations pre-existed already at baseline at low variant allele frequencies (VAFs) of 1–3 %. Patients with these pre-existing mutations had similar responses to pirtobrutinib, with an ORR of 75 %.
Optimizing outcomes on pirtobrutinib
Varsha Gandhi, MD, Anderson Cancer Center, Houston, Texas, reported research efforts into strategies to improve clinical results in pirtobrutinib-treated patients. At the MD Anderson Cancer Center, 14 of 23 evaluable patients with relapsed/refractory CLL who received pirtobrutinib in the BRUIN study developed progression. In this group, different types of second-site mutations prevailed. Only two individuals had BTK wildtype, and the C481 clone was eradicated in almost all cases. The researchers sought to identify genomic features at baseline that were associated with maintenance of the response to pirtobrutinib. According to this, risk factors for progression on pirtobrutinib included relapsed/refractory status after previous covalent BTK inhibition (rather than intolerance to these agents) and the presence of BTK mutations at baseline, complex karyotype and bulky lymph nodes. As Dr. Gandhi pointed out, this observation needs to be validated in the larger BRUIN cohort. Nevertheless, it implies that pirtobrutinib should preferably be used in previously untreated CLL patients.
Pharmacologic profiling was performed to develop a sequencing strategy during pirtobrutinib therapy, while the cells are sensitive to treatment, to achieve deeper responses. Mononuclear cells obtained in the course of treatment were incubated with ibrutinib, acalabrutinib, venetoclax and their combinations, and their viability was assessed. Indeed, after ≥ 10 cycles of pirtobrutinib treatment, the cells showed increased sensitivity to targeted agents alone and in combination.
Moreover, post-progression therapeutic intensification options were investigated based on pharmacological and genetic or genomic profiling. Characterization of cells showed that the chemokines CCL3 and CCL4 were increased at the time of progression, which indicated activation of the B-cell receptor pathway. Also, phosphorylation of BTK, ERK and NF-κβ increased, as did BCL-2 and MCL-1 protein levels. These pirtobrutinib-resistant cells proved sensitive to ibrutinib as well as to doublets and triplets containing BCL-2–, MCL-1– and BTK-targeted agents. This was confirmed in vivo. Eleven patients were sensitive to the combinations regardless of their genomic background, with the exception of BCL-2 mutations that abrogated the treatment effect. Overall, these results hint at options for treatment sequencing, intensification and reuse of covalent BTK inhibition. Dr. Gandhi emphasized that an unmet need persists in patients with double-refractory CLL harboring second-site BTK and BCL-2 mutations.
Heterogeneity of BCL-2 resistance
Insights into mechanisms of resistance to BCL-2–targeted treatment were reported by Rachel Thijssen, PhD, The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia and Amsterdam UMC, the Netherlands. CLL cells from relapsed patients have shown to be intrinsically less sensitive to venetoclax [9]. Among intrinsic mechanisms, the BCL-2 Gly101Val mutation that reduces binding affinity of venetoclax to BCL-2 in the CLL cell was identified [10]. However, this is subclonal and is found only in a subset of patients progressing on venetoclax.
The researchers applied single-cell rapid capture hybridization sequencing (scRaCH-seq), a novel multi-omics approach that allows for the assessment of transcriptomes and genotypes of single cells. According to this, CLL cells at progression have different transcriptome profiles, with various mechanisms of resistance in the same patient sample [9]. Intrinsic resistance was found to be driven by high MCL-1 and NF-κB activation with ongoing venetoclax therapy. Most of the relapse samples showed an increased expression of MCL-1, which was strongly correlated with that of REL, RELB and NFKB2 (Figure 2). Moreover, a universal activation of NF-kB and increased MCL-1 expression were observed in circulating resistant tumor cells. These characteristics dissipated after venetoclax treatment had been discontinued, and according to the ex vivo sensitivity assay, the cells were again sensitive to the BCL-2 inhibitor.
Regarding extrinsic resistance mechanisms, a shift was seen in the expression of BCL-2 family members. CLL cells in lymph nodes had different transcriptome profiles than CLL cells in the peripheral blood. Overall, the researchers observed a high degree of interpatient and intrapatient heterogeneity at venetoclax relapse, both intrinsically and extrinsically. The significance of MCL-1 and NF-κB activation with ongoing venetoclax therapy underscores the importance of time-limited treatment. Considering the findings regarding extrinsic resistance, combinations of venetoclax with agents preventing microenvironment support appear warranted. The question of whether fixed-duration treatment with venetoclax plus BTK inhibition prevents drug resistance remains to be answered.
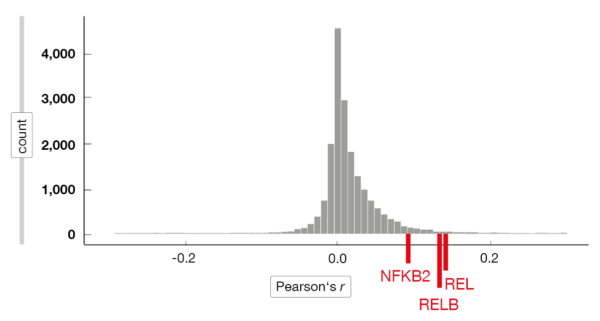
Figure 2: Correlation between MCL-1 and other genes
Mutation patterns in the real world
Real-world information is required on the patterns of mutations during BTK inhibitor treatment, especially with non-covalent agents. Kiyomi Mashima, MD, PhD, Dana-Farber Cancer Institute and Harvard Medical School, Boston, USA, presented the results of a retrospective cohort analysis of CLL patients treated with ibrutinib, acalabrutinib or pirtobrutinib at the Dana-Farber Cancer Institute between 2014 and 2022. The scientists included 118 clinical sequencing reports from 85 patients who were divided into two groups, one undergoing NGS at the time of disease progression (PD; n = 36) and another without progression (non-progressors, NP) in whom NGS was performed during response (n = 49). Sixty and 58 of the sequencing reports were obtained from the PD and NP groups, respectively.
Mutations that were new or had increased VAF were significantly more common among PD vs. NP patients (p < 0.001). Significant differences were found for TP53, BTK, SF3B1, PLCG2 and NOTCH2. Only patients from the NP group did not develop any mutations. The analysis revealed six different types of C481 mutations. Interestingly, L528W mutations arose on both ibrutinib (2/7) and pirtobrutinib (1/6) treatment. Four of six patients progressing on pirtobrutinib had T474 mutations.
Paired sequencing results before and during BTK inhibitor therapy were obtained. All of the patients in the PD group exhibited at least one new or increased BTK, PLCG2 or TP53 mutation. In contrast, in the NP group, none of these mutations newly appeared or increased. Four pre-existing TP53 mutations and six pre-existing NOTCH1 mutations decreased or completely disappeared during BTK inhibitor treatment in the NP group. Overall, the enrichment of RAS/RAF/MAPK pathway mutations in the PD group suggests that these may be related to BTK inhibitor resistance beyond BTK, PLCG2 and TP53 mutations.
What can be achieved with BTK degraders?
Kinase inhibition might not suffice to overcome resistance to BTK inhibitors as some BTK mutations carry a scaffold function that cannot be surmounted by tyrosine kinase inhibitors [11]. Therefore, targeted protein degradation has emerged as a strategy to circumvent acquired resistance to BTK inhibitors. Proteolysis-targeted chimeras (PROTAC) are a novel class of agents that enable catalytic ubiquitination and proteasomal degradation of different targets. Alexey V. Danilov, MD, PhD, Lymphoma Center, City of Hope, Irwindale, USA, explained that kinases such as BTK, BRAF or KRAS can be targeted, although this basically applies to any type of protein.
Multiple covalent and non-covalent BTK degraders are currently in preclinical development. An example is NRX-0492 that degrades BTK, IL2-inducible T-cell kinase (ITK) and IKAROS family zinc finger (IKZF), thus inhibiting B-cell receptor signaling [12]. Importantly, BTK degradation overcomes BTK resistance mutations. In a preclinical resistance model, NX-2127 reduced proliferation and induced apoptosis (Figure 3) while ibrutinib failed to do so [13-15]. NX-2127 and NX-5948 induce BTK degradation by recruiting cereblon that was originally recognized for its modulation of the immune system. However, only NX-2127 was shown to downregulate the transcription factors Aiolos and Ikaros, which indicates an immunomodulatory function of this compound [16]. NX-2127, but not NX-5948, enhanced synapse formation to levels comparable to those of lenalidomide. Whether this is clinically important in terms of efficacy and toxicity remains to be determined.
The BTK degraders NX-2127, NX-5948, BGB-16673, AC0676 and ABBV-101 are being tested in clinical trials. Early data from the phase I NX-2127-001 study investigating NX-2127 in patients with relapsed/refractory CLL and B-cell malignancies have been presented at ASH 2022 [17]. All of the 23 CLL patients were heavily pretreated and were refractory to prior BTK inhibitor therapy, with most being double-refractory to both BTK and BCL-2 inhibitors. The adverse event (AE) profile was generally consistent with previous reports for BTK-targeted therapies. NX-2127 showed clinically meaningful efficacy, with an ORR of 33 % and durable responses in some patients. Importantly, responses were noted in patients double-refractory to BTK/BCL-2 inhibitors as well as in those who had progressed on a non-covalent BTK inhibitor. In vivo BTK degradation was significant and was essentially independent of the dose level.
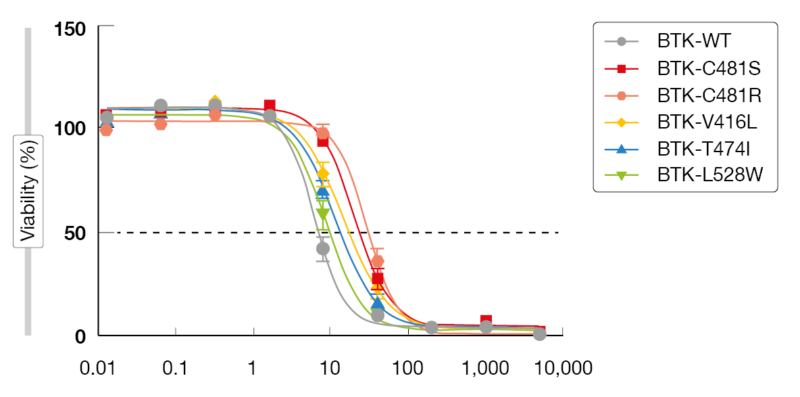
Figure 3: Activity of NX-2127 against BTK inhibitor resistance mutations
Zanubrutinib in patients intolerant of other BTKi
The ongoing, single-arm, phase II BGB-3111-215 study has demonstrated efficacy and tolerability of zanubrutinib in patients with B-cell malignancies who are intolerant of ibrutinib and/or acalabrutinib [18]. Preliminary longer-term findings for patients with CLL or small lymphocytic lymphoma were presented at iwCLL 2023 by Shadman et al. [19]. Overall, 61 individuals received zanubrutinib; 44 and 17 of these were ibrutinib-intolerant and acalabrutinib-intolerant, respectively. In the latter group, eight patients were intolerant of both agents. At the time of the analysis, approximately 70 % in each cohort were still on treatment.
The data showed that AEs that had caused the patients to discontinue their previous BTK inhibitor therapy were unlikely to recur on zanubrutinib treatment. Indeed, 68.4 % of ibrutinib-intolerance AEs and 71.4 % of acalabrutinib-intolerance AEs did not recur (Figure 4). No intolerance AEs recurred at a higher severity. Sixty-two percent of patients with ibrutinib intolerance and 70.6 % of those with acalabrutinib intolerance did not experience recurrence of any of their intolerance events. Grade ≥ 3 AEs emerging on zanubrutinib treatment were reported in 50.8 % of patients, with neutropenia (11.5 %), COVID-19 (6.6 %) and pneumonia (6.6 %) representing the most common events.
In terms of efficacy of zanubrutinib, the analysis showed an ORR of 71.9 % and a disease control rate of 94.7 %. At 12 months, 88.3 % of patients were progression-free. As the authors noted, the high disease control rate suggests that patients who were intolerant of ibrutinib or acalabrutinib are likely to benefit from switching to zanubrutinib. Overall, zanubrutinib appears to be a viable option for patients who are intolerant of ibrutinib or acalabrutinib.
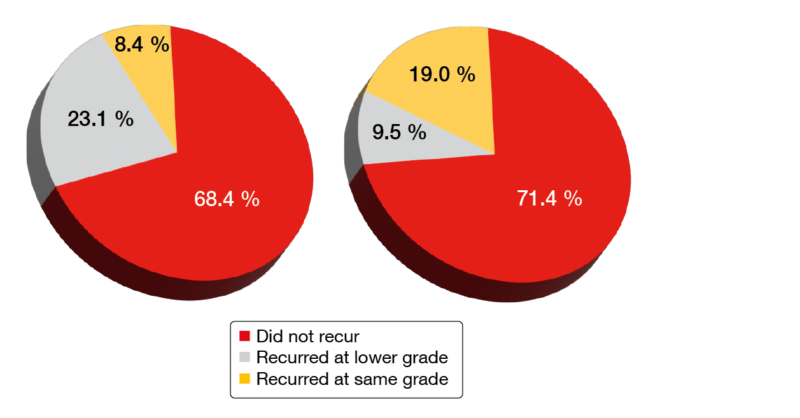
Figure 4: Recurrence of ibrutinib-intolerance events (left) and acalabrutinib-intolerance events (right) on zanubrutinib treatment
Assessment of evolutionary characteristics
Zhu et al. used multi-omics to assess clonal evolutionary characteristics in ten retrospectively identified CLL patients who had developed zanubrutinib resistance [20]. This included two patients with Richter’s transformation. Deep targeted-gene NGS covering BTK (exons 1–19) and PLCG2 (exons 1–33), as well as high sensitivity droplet digital PCR (ddPCR) were used to assess serial samples. Moreover, the scientists performed single-cell RNA sequencing of matching peripheral blood and lymph node samples in three patients who showed progressive lymphadenopathy.
BTK Cys481 and Leu528 mutations were identified as the two main BTK resistance mutations. ddPCR was performed in the samples of four patients with undetectable BTK mutation according to NGS. The Cys481Ser mutation was found in two cases, which suggested spatial clonal heterogeneity in zanubrutinib resistance. As the authors noted, deep targeted-gene NGS and ddPCR should be used comprehensively to evaluate the emergence of resistant clones in light of spatial heterogeneity and clonal evolution. Patients with Richter’s transformation showed the highest upregulation of the MYC, OXPHOS and G2M pathways, as well as downregulation of BCR signaling. Furthermore, upregulation of MCL-1 in this group indicated an underlying mechanism leading to insensitivity to venetoclax treatment following zanubrutinib resistance.
Source: Sessions “Mechanisms of resistance” and “Future directions for the treatment of relapsed/refractory CLL”, iwCLL 2023, 8th October 2023, Boston, USA
REFERENCES
- Johnson AR et al., Battling Btk mutants with noncovalent inhibitors that overcome Cys481 and Thr474 mutations. ACS Chem Biol 2016; 11(10): 2897-2907
- Lin DY, Andreotti AH, Structure of BTK kinase domain with the second-generation inhibitors acalabrutinib and tirabrutinib. PLoS One 2023; 18(8): e0290872
- Ahn IE et al., Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood 2017; 129(11): 1469-1479
- Ahn IE et al., Ibrutinib for chronic lymphocytic leukemia with TP53 alterations. N Engl J Med 2020; 383(5): 498-500
- Burger JA et al., Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Comm 2016; 7: 11589
- Kadri S et al., Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv 2017; 1(12): 715-727
- Bulian P et al., CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J Clin Oncol 2014; 32(9): 897-904
- Alsadhan A et al., CD49d expression identifies a biologically distinct subtype of chronic lymphocytic leukemia with inferior progression-free survival on BTK inhibitor therapy. Clin Cancer Res 2023; 29(18): 3612-3621
- Thijssen R et al., Single-cell multiomics reveal the scale of multilayered adaptations enabling CLL relapse during venetoclax therapy. Blood 2022; 140(20): 2127-2141
- Blombery P et al., Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov 2019; 9(3): 342-353
- Wang et al., Mechanisms of resistance to noncovalent Bruton’s tyrosine kinase inhibitors. N Engl J Med 2022; 386(8): 735-743
- Zhang D et al., NRX-0492 degrades wild-type and C481 mutant BTK and demonstrates in vivo activity in CLL patient-derived xenografts. Blood 2023; 141(13): 1584-1596
- Abdel-Wahab O et al., Kinase dead BTK mutations confer resistance to covalent and noncovalent BTK inhibitors but are susceptible to clinical stage BTK degraders. ASH 2022, abstract 750
- Noviski M et al., NX-5948 promotes selective, sub-nanomolar degradation of inhibitor-resistant BTK mutants. AACR 2023, abstract 2850
- Noviski M et al., NX-5948 and NX-2127 potently degrade a broad array of clinically-relevant BTK mutants that display resistance to inhibitors and other BTK degraders. iwCLL 2023, poster 2020
- Huynh T et al., NX-2127 and NX-5948, two clinical stage cereblon-recruiting BTK degraders, facilitate T-cell functionality in CLL. iwCLL 2023, poster 1050
- Mato AR et al., NX-2127-001, a first-in-human trial of NX-2127, a Bruton’s tyrosine kinase-targeted protein degrader, in patients with relapsed or refractory chronic lymphocytic leukemia and B-cell malignancies. Blood 2022; 140 (Supplement 1): 2329-2332
- Shadman M et al., Zanubrutinib in patients with previously treated B-cell malignancies intolerant of previous Bruton tyrosine kinase inhibitors in the USA: a phase 2, open-label, single-arm study. Lancet Haematol 2023; 10(1): e35-e45
- Shadman M et al., A phase 2 study of zanubrutinib in previously treated B-cell malignancies intolerant to ibrutinib and/or acalabrutinib: preliminary results for patients with CLL/SLL. iwCLL 2023, poster 2009
- Zhu H et al., Integrating multi-omics to reveal the heterogeneous clonal evolutionary characteristics in CLL patients with zanubrutinib resistance. iwCLL 2023, poster 1102
© 2023 Springer-Verlag GmbH, Impressum
More posts
Long-term results and other findings from clinical trials
Long-term results and other findings from clinical trials MURANO: 7-year data and retr
Preface – iwCLL 2023
Preface – iwCLL 2023 © author's own – Kiyomi Mashima MD, PhD, Medical Oncology, CLL C