

Innovative agents in marginal zone lymphoma and other B-cell malignancies
Relapsed/refractory MZL: sonrotoclax
Relapses are common in patients with marginal zone lymphoma (MZL), and sequential therapy is often necessary. At ASH 2023, Tedeschi et al. reported findings for 22 patients with relapsed/refractory MZL who received the oral second-generation Bcl-2 inhibitor sonrotoclax at different dose levels (i.e., 40 mg, 160 mg, 320 mg, 640 mg OD) in the first-in-human, phase I, multicenter BGB-11417-101 study [1]. Dose expansion started with the 640 mg dose; the 320 mg dose was expanded later to include 10 additional patients.
Ten participants received sonrotoclax 640 mg. Even at this dose level, the maximum tolerated dose was not reached, although pyrexia was more common with the 640 mg dose than with all other doses (40 % vs. 25 %). This also applied to constipation (30 % vs. 17 %), diarrhea (30 % vs. 8 %), and headache (30 % vs. 8 %). No clinical tumor lysis syndrome (TLS) occurred, while two transitory cases of laboratory TLS emerged in patients with high baseline levels of circulating cells, including a patient with a very large spleen that significantly decreased in size with the first dose. Dose interruptions were called for in three patients due to adverse events (AEs), and one patient had to discontinue treatment.
In the efficacy-evaluable cohort, 70 % responded to sonrotoclax 640 mg, with 40 % achieving complete response (CR). Responses proved durable in the 640 mg group; at a median follow-up of 8.7 months, 6 of 10 patients were continuing on treatment. Taken together, sonrotoclax demonstrated promising single-agent activity in the setting of relapsed/refractory MZL.
Non-covalent BTK inhibition with pirtobrutinib
Another option under investigation in relapsed/refractory MZL is the non-covalent BTK inhibitor pirtobrutinib. The phase I/II BRUIN study evaluated pirtobrutinib in 36 patients with MZL after a median of three prior systemic treatment lines. Covalent BTK inhibition (cBTKi) had been administered in 72 % of patients, and all of them had received anti-CD20 antibodies. Progression had been the reason for discontinuation of cBTKi therapy in 77 %.
In this heavily pretreated cohort, pirtobrutinib showed encouraging efficacy with an overall response rate (ORR) of 50.0 % in the total cohort; in patients with and without previous cBTKi therapy, the ORRs were 46.2 % and 60.0 %, respectively (Table) [2]. Responses lasted for a median of 12.7 months, and median progression-free survival was 16.5 months. For both of these endpoints, the results were improved in cBTKi-naïve patients compared to the cBTKi-pretreated group. Median overall survival had not been reached in any of these cohorts. At 18 months, 81.8 % of all patients were alive, and 46.6 % were progression-free.
Pirtobrutinib was well tolerated. Treatment-related adverse events (TRAEs) required discontinuation and dose reduction in 5.6 % and 11.1 %, respectively. Among TRAEs of interest, bruising and rash were most common (any grade, 25.0 % and 19.4 %, respectively). No grade 3/4 TRAEs occurred with the exception of hypertension (2.8 %). The authors concluded that pirtobrutinib might be a promising chemotherapy-free option after cBTKi pretreatment in patients with relapsed/refractory MZL.
BTK degrader BGB-16673
Disease progression in BTK-inhibitor–treated patients with B-cell malignancies can be due to resistance mutations within BTK that arise on both covalent and non-covalent agents [3, 4]. Consequently, there is a need for new approaches to address the shortcomings of existing BTK inhibitors. The BTK degrader BGB-16673 has been shown to degrade both wild-type BTK and covalent as well as non-covalent BTK-inhibitor–resistant mutant proteins such as V416L, M437R, T474I, C481S, thus inducing tumor suppression [5, 6].
Seymour et al. presented preliminary safety and efficacy results for 50 patients enrolled in the ongoing BGB-16673-101 study that is testing BGB-16673 in patients with relapsed or refractory B-cell malignancies including MZL, follicular lymphoma (FL), mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), Waldenström macroglobulinemia (WM), diffuse large B-cell lymphoma (DLBCL), and Richter transformation [7]. Prior to inclusion, patients had been treated with ≥ 2 therapies and had received a covalent BTK inhibitor if this was locally approved for their disease. BGB-16673 was administered at five dose levels ranging from 50 mg to 500 mg.
The agent resulted in meaningful responses, with an ORR of 57 % at all doses and a median time to first response of 2.76 months. The disease control rate was 75 %. Seven out of 10 patients with CLL/SLL, all of whom were pretreated with covalent BTK inhibitors, responded to the treatment. The disease control rate in this group was 90 %. Patients with MCL, MZL or WM achieved responses and disease control in 56 % and 75 %, respectively. Furthermore, substantial reductions in BTK protein levels were observed in the peripheral blood and tumor tissue, which is a proof of concept of a strong on-target effect.
The safety profile of BGB-16673 appeared tolerable. Grade 3 rash was reported as the only dose-limiting toxicity, occurring in a single patient. Two patients discontinued BGB-16673 due to treatment-emergent AEs (TEAEs). No atrial fibrillation or hypertension had occurred at the time of data cutoff. Phase II dose expansions within the BGB-16673-101 study are planned for patients with CLL/SLL and MCL.
NX-2127: immunomodulation plus BTK degradation
Preclinical and clinical evidence suggests that modulation of cereblon to degrade Ikaros family proteins might synergize with BTK inhibition in non-Hodgkin lymphoma (NHL) [8]. NX-2127 is an oral, first-in-class small molecule that combines BTK degradation with the immunomodulatory activity of a degrader for the transcription factor Ikaros [9]. Compared with classical BTK inhibitors, NX-2127 is expected to exert superior efficacy by overcoming BTK-mutation–driven resistance. The first-in-human, phase Ia/Ib NX-2127-001 study is currently assessing NX-2127 in patients with advanced B-cell malignancies. Updated safety and efficacy data were reported for a total of 54 patients with relapsed/refractory DLBCL, MCL, MZL, WM, FL and CLL/SLL at ASH 2023 [10]. In the overall population, the median number of prior treatment lines was 4. Patients were predominantly elderly, had received multiple lines of targeted agents and showed acquired mutations associated with drug resistance.
NX-2127 demonstrated a manageable safety profile that was consistent with previous reports for BTK-targeted and immunomodulatory compounds. The most common TEAEs included neutropenia (any grade, 46.3 %; grade ≥ 3, 42.6 %), fatigue (46.3 %; no grade ≥ 3 events), and hypertension (33.3 %; 14.8 %). Atrial fibrillation was observed in 11.1 % of patients, with 5.6 % experiencing grade ≥ 3 events. Two patients developed dose-limiting toxicities at the 300 mg dose level, and NX-2127 had to be discontinued due to TEAEs in 13 patients.
The treatment gave rise to rapid, robust and sustained degradation of BTK (Figure) in all patients regardless of their absolute BTK starting level, tumor type, or NX-2127 dose level. Consistent with the immunomodulatory activity of NX-2127, biologically relevant degradation of Ikaros was observed. In the group with NHL and WM (n = 17), two individuals each showed complete and partial responses. Among 27 patients with CLL/SLL, 11 (40.7 %) developed partial responses with and without lymphocytosis. Twelve patients (44.4 %) had stable disease at the time of data cutoff. Responses were seen in double- and triple-exposed individuals. Eight patients had been on treatment for > 12 months at the time of the analysis, and treatment was ongoing in 13 individuals. Dose-expansion cohorts of patients with NHL have been initiated at the 300 mg dose level.
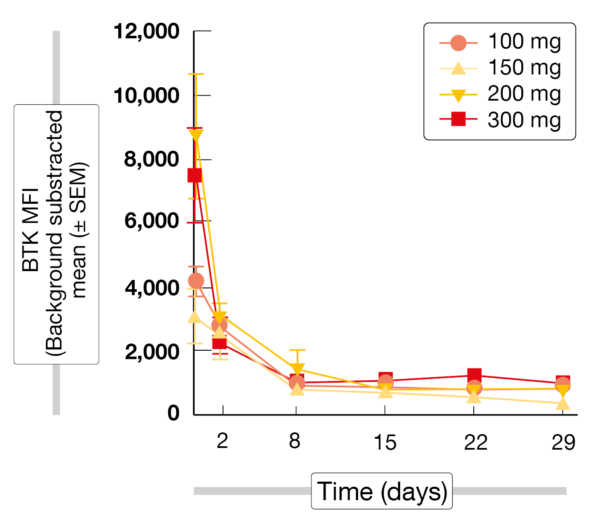
Figure: BTK degradation with NX-2127 at dose levels of 100-300 mg
REFERENCES
- Tedeschi A et al., Monotherapy with second-generation BCL2 inhibitor sonrotoclax (BGB-11417) is well tolerated with high response rates in patients with relapsed/refractory marginal zone lymphoma: Data from an ongoing phase 1 study. ASH 2023, abstract 3032
- Patel K et al., Pirtobrutinib, a highly selective, non-covalent (reversible) BTK inhibitor in relapsed/refractory marginal zone lymphoma: results from phase 1/2 BRUIN study. ASH 2023, abstract 1660
- Woyach JA et al., Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med 2014; 370(24): 2286-2294
- Wang E et al., Mechanisms of resistance to noncovalent Bruton’s tyrosine kinase inhibitors. N Engl J Med 2022; 386(8): 735-743
- Feng X et al., Bruton tyrosine kinase protein degrader BGB-16673 is less apt to cause, and able to overcome variable BTK resistance mutations compared to other BTK inhibitors. EHA 2023, abstract P1239
- Wang H et al., BGB-16673, a BTK degrader, overcomes on-target resistance from BTK inhibitors and presents sustainable long-term tumor regression in lymphoma xenograft models. EHA 2023, abstract P1219
- Seymour JF et al., First results from a phase 1, first-in-human study of the Bruton Tyrosine kinase degrader BGB-16673 in patients with relapsed or refractory B-cell malignancies. ASH 2023, abstract 4401
- Montoya S et al., Efficacy of pirtobrutinib in covalent BTK-inhibitor pre-treated relapsed/refractory CLL/SLL: additional patients and extended follow-up from the phase 1/2 BRUIN study. Blood 2022; 140(Suppl 1): 1811-1813
- Noviski M et al., Concurrent degradation of BTK and IMiD neosubstrates by NX-2127 enhances multiple mechanisms of tumor killing. Cancer Res 2022; 82 (Suppl 12): 1128
- Danilov A et al., A first-in-human phase 1 trial of NX-2127, a first-in-class Bruton’s tyrosine kinase dual-targeted protein degrader with immunomodulatory activity, in patients with relapsed/refractory B-cell malignancies. ASH 2023, abstract 4463
© 2024 Springer-Verlag GmbH, Impressum